T003 · Molecular filtering: unwanted substructures¶
Note: This talktorial is a part of TeachOpenCADD, a platform that aims to teach domain-specific skills and to provide pipeline templates as starting points for research projects.
Authors:
Maximilian Driller, CADD seminar, 2017, Charité/FU Berlin
Sandra Krüger, CADD seminar, 2018, Charité/FU Berlin
Talktorial T003: This talktorial is part of the TeachOpenCADD pipeline described in the first TeachOpenCADD publication (J. Cheminform. (2019), 11, 1-7), comprising of talktorials T001-T010.
Aim of this talktorial¶
There are some substructures we prefer not to include into our screening library. In this talktorial, we learn about different types of such unwanted substructures and how to find, highlight and remove them with RDKit.
Contents in Theory¶
Unwanted substructures
Pan Assay Interference Compounds (PAINS)
Contents in Practical¶
Load and visualize data
Filter for PAINS
Filter for unwanted substructures
Highlight substructures
Substructure statistics
References¶
Pan Assay Interference compounds (wikipedia, J. Med. Chem. (2010), 53, 2719-2740)
Unwanted substructures according to Brenk et al. (Chem. Med. Chem. (2008), 3, 435-44)
Inspired by a Teach-Discover-Treat tutorial (repository)
RDKit (repository, documentation)
[1]:
import sys
if "google.colab" in sys.modules:
%pip install teachopencadd --no-deps -q
!teachopencadd -d 3
%pip uninstall teachopencadd -y -q
%pip install -qr requirements.txt
Theory¶
Unwanted substructures¶
Substructures can be unfavorable, e.g., because they are toxic or reactive, due to unfavorable pharmacokinetic properties, or because they likely interfere with certain assays. Nowadays, drug discovery campaigns often involve high throughput screening. Filtering unwanted substructures can support assembling more efficient screening libraries, which can save time and resources.
Brenk et al. (Chem. Med. Chem. (2008), 3, 435-44) have assembled a list of unfavorable substructures to filter their libraries used to screen for compounds to treat neglected diseases. Examples of such unwanted features are nitro groups (mutagenic), sulfates and phosphates (likely resulting in unfavorable pharmacokinetic properties), 2-halopyridines and thiols (reactive). This list of undesired substructures was published in the above mentioned paper and will be used in the practical part of this talktorial.
Pan Assay Interference Compounds (PAINS)¶
PAINS are compounds that often occur as hits in HTS even though they actually are false positives. PAINS show activity at numerous targets rather than one specific target. Such behavior results from unspecific binding or interaction with assay components. Baell et al. (J. Med. Chem. (2010), 53, 2719-2740) focused on substructures interfering in assay signaling. They described substructures which can help to identify such PAINS and provided a list which can be used for substructure filtering.
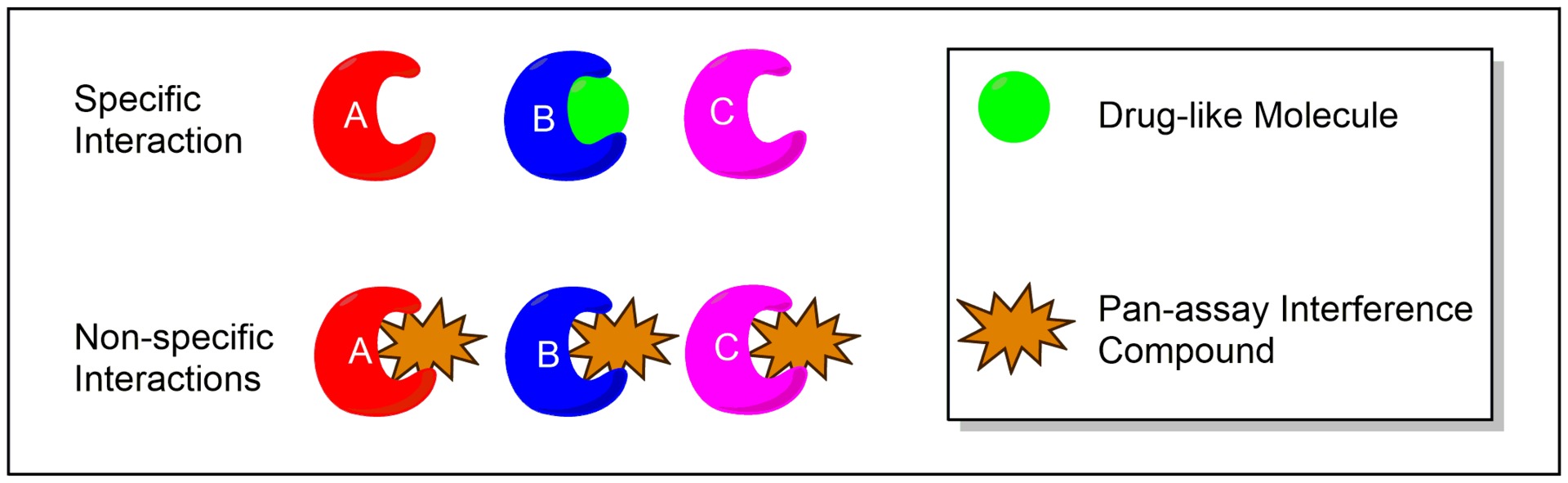
Figure 1: Specific and unspecific binding in the context of PAINS. Figure taken from Wikipedia.
Practical¶
Load and visualize data¶
First, we import the required libraries, load our filtered dataset from Talktorial T002 and draw the first molecules.
[2]:
from pathlib import Path
import pandas as pd
from tqdm.auto import tqdm
from rdkit import Chem
from rdkit.Chem import PandasTools
from rdkit.Chem.FilterCatalog import FilterCatalog, FilterCatalogParams
/opt/miniconda3/envs/T003_3120/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
<frozen importlib._bootstrap>:488: RuntimeWarning: to-Python converter for boost::shared_ptr<RDKit::FilterHierarchyMatcher> already registered; second conversion method ignored.
[3]:
# define paths
HERE = Path(_dh[-1])
DATA = HERE / "data"
[4]:
# load data from Talktorial T2
egfr_data = pd.read_csv(DATA / "EGFR_compounds_lipinski.csv", index_col=0)
# Drop unnecessary information
print("Dataframe shape:", egfr_data.shape)
egfr_data.drop(columns=["molecular_weight", "n_hbd", "n_hba", "logp"], inplace=True)
egfr_data.head()
Dataframe shape: (4635, 10)
[4]:
| molecule_chembl_id | IC50 | units | smiles | pIC50 | ro5_fulfilled | |
|---|---|---|---|---|---|---|
| 0 | CHEMBL63786 | 0.003 | nM | Brc1cccc(Nc2ncnc3cc4ccccc4cc23)c1 | 11.522879 | True |
| 1 | CHEMBL35820 | 0.006 | nM | CCOc1cc2ncnc(Nc3cccc(Br)c3)c2cc1OCC | 11.221849 | True |
| 2 | CHEMBL53711 | 0.006 | nM | CN(C)c1cc2c(Nc3cccc(Br)c3)ncnc2cn1 | 11.221849 | True |
| 3 | CHEMBL66031 | 0.008 | nM | Brc1cccc(Nc2ncnc3cc4[nH]cnc4cc23)c1 | 11.096910 | True |
| 4 | CHEMBL53753 | 0.008 | nM | CNc1cc2c(Nc3cccc(Br)c3)ncnc2cn1 | 11.096910 | True |
[5]:
# Add molecule column
PandasTools.AddMoleculeColumnToFrame(egfr_data, smilesCol="smiles")
# Draw first 3 molecules
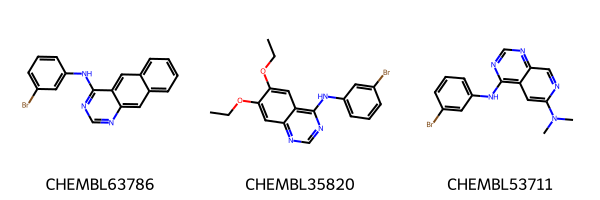
Chem.Draw.MolsToGridImage(
list(egfr_data.head(3).ROMol),
legends=list(egfr_data.head(3).molecule_chembl_id),
)
[5]:
Filter for PAINS¶
The PAINS filter is already implemented in RDKit (documentation). Such pre-defined filters can be applied via the FilterCatalog class. Let’s learn how it can be used.
[6]:
# initialize filter
params = FilterCatalogParams()
params.AddCatalog(FilterCatalogParams.FilterCatalogs.PAINS)
catalog = FilterCatalog(params)
[7]:
# search for PAINS
matches = []
clean = []
for index, row in tqdm(egfr_data.iterrows(), total=egfr_data.shape[0]):
molecule = Chem.MolFromSmiles(row.smiles)
entry = catalog.GetFirstMatch(molecule) # Get the first matching PAINS
if entry is not None:
# store PAINS information
matches.append(
{
"chembl_id": row.molecule_chembl_id,
"rdkit_molecule": molecule,
"pains": entry.GetDescription().capitalize(),
}
)
else:
# collect indices of molecules without PAINS
clean.append(index)
matches = pd.DataFrame(matches)
egfr_data = egfr_data.loc[clean] # keep molecules without PAINS
100%|██████████| 4635/4635 [00:18<00:00, 246.19it/s]
[8]:
# NBVAL_CHECK_OUTPUT
print(f"Number of compounds with PAINS: {len(matches)}")
print(f"Number of compounds without PAINS: {len(egfr_data)}")
Number of compounds with PAINS: 408
Number of compounds without PAINS: 4227
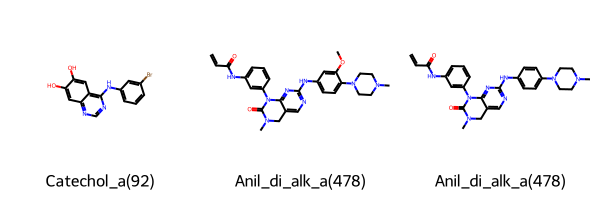
Let’s have a look at the first 3 identified PAINS.
[9]:
Chem.Draw.MolsToGridImage(
list(matches.head(3).rdkit_molecule),
legends=list(matches.head(3)["pains"]),
)
[9]:
Filter and highlight unwanted substructures¶
Some lists of unwanted substructures, like PAINS, are already implemented in RDKit. However, it is also possible to use an external list and get the substructure matches manually. Here, we use the list provided in the supporting information from Brenk et al. (Chem. Med. Chem. (2008), 3, 535-44).
[10]:
substructures = pd.read_csv(DATA / "unwanted_substructures.csv", sep=r"\s+")
substructures["rdkit_molecule"] = substructures.smarts.apply(Chem.MolFromSmarts)
print("Number of unwanted substructures in collection:", len(substructures))
# NBVAL_CHECK_OUTPUT
Number of unwanted substructures in collection: 105
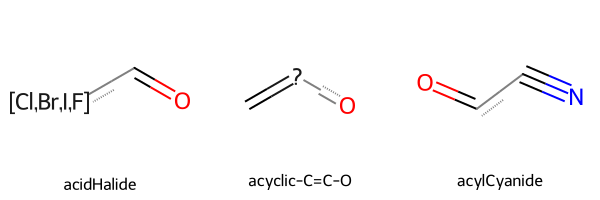
Let’s have a look at a few substructures.
[11]:
Chem.Draw.MolsToGridImage(
mols=substructures.rdkit_molecule.tolist()[2:5],
legends=substructures.name.tolist()[2:5],
)
[11]:
Search our filtered dataframe for matches with these unwanted substructures.
[12]:
# search for unwanted substructure
matches = []
clean = []
for index, row in tqdm(egfr_data.iterrows(), total=egfr_data.shape[0]):
molecule = Chem.MolFromSmiles(row.smiles)
match = False
for _, substructure in substructures.iterrows():
if molecule.HasSubstructMatch(substructure.rdkit_molecule):
matches.append(
{
"chembl_id": row.molecule_chembl_id,
"rdkit_molecule": molecule,
"substructure": substructure.rdkit_molecule,
"substructure_name": substructure["name"],
}
)
match = True
if not match:
clean.append(index)
matches = pd.DataFrame(matches)
egfr_data = egfr_data.loc[clean]
100%|██████████| 4227/4227 [00:31<00:00, 134.35it/s]
[13]:
# NBVAL_CHECK_OUTPUT
print(f"Number of found unwanted substructure: {len(matches)}")
print(f"Number of compounds without unwanted substructure: {len(egfr_data)}")
Number of found unwanted substructure: 3234
Number of compounds without unwanted substructure: 2088
Highlight substructures¶
Let’s have a look at the first 3 identified unwanted substructures. Since we have access to the underlying SMARTS patterns we can highlight the substructures within the RDKit molecules.
[14]:
to_highlight = [
row.rdkit_molecule.GetSubstructMatch(row.substructure) for _, row in matches.head(3).iterrows()
]
Chem.Draw.MolsToGridImage(
list(matches.head(3).rdkit_molecule),
highlightAtomLists=to_highlight,
legends=list(matches.head(3).substructure_name),
)
[14]:
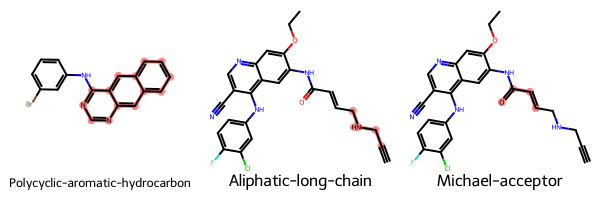
Substructure statistics¶
Finally, we want to find the most frequent substructure found in our data set. The Pandas DataFrame provides convenient methods to group containing data and to retrieve group sizes.
[15]:
# NBVAL_CHECK_OUTPUT
groups = matches.groupby("substructure_name")
group_frequencies = groups.size()
group_frequencies.sort_values(ascending=False, inplace=True)
group_frequencies.head(10)
[15]:
substructure_name
Michael-acceptor 1113
Aliphatic-long-chain 489
Oxygen-nitrogen-single-bond 367
triple-bond 252
nitro-group 177
imine 150
Thiocarbonyl-group 114
aniline 64
halogenated-ring 62
conjugated-nitrile-group 59
dtype: int64
Discussion¶
In this talktorial we learned two possibilities to perform a search for unwanted substructures with RDKit:
The
FilterCatalogclass can be used to search for predefined collections of substructures, e.g., PAINS.The
HasSubstructMatch()function to perform manual substructure searches.
Actually, PAINS filtering could also be implemented via manual substructure searches with HasSubstructMatch(). Furthermore, the substructures defined by Brenk et al. (Chem. Med. Chem. (2008), 3, 535-44) are already implemented as a FilterCatalog. Additional pre-defined collections can be found in the RDKit documentation.
So far, we have been using the HasSubstructMatch() function, which only yields one match per compound. With the GetSubstructMatches() function (documentation) we have the opportunity to identify all occurrences of a particular substructure in a compound. In case of PAINS, we have only looked at the first match per molecule (GetFirstMatch()). If we simply want to filter out all PAINS this is enough. However, we could also
use GetMatches() in order to see all critical substructures of a molecule.
Detected substructures can be handled in two different fashions:
Either, the substructure search is applied as a filter and the compounds are excluded from further testing to save time and money.
Or, they can be used as warnings, since ~5 % of FDA-approved drugs were found to contain PAINS (ACS. Chem. Biol. (2018), 13, 36-44). In this case experts can judge manually, if an identified substructure is critical or not.
Quiz¶
Why should we consider removing “PAINS” from a screening library? What is the issue with these compounds?
Can you find situations when some unwanted substructures would not need to be removed?
How are the substructures we used in this tutorial encoded?