T014 · Binding site detection¶
Note: This talktorial is a part of TeachOpenCADD, a platform that aims to teach domain-specific skills and to provide pipeline templates as starting points for research projects.
Authors:
Adapted from Abishek Laxmanan Ravi Shankar, 2019, internship at Volkamer lab
Andrea Volkamer, 2020/21, Volkamer lab, Charité
Dominique Sydow, 2020/21, Volkamer lab, Charité
Aim of this talktorial¶
The binding site of a protein is the key to its function. In this talktorial, we introduce the concepts of computational binding site detection tools using DoGSiteScorer from the protein.plus web server, exemplified on an EGFR structure. Additionally, we compare the results to the pre-defined KLIFS binding site by calculating the percentage of residues in accordance between the two sets.
Contents in Theory¶
Protein binding sites
Binding site detection
Methods overview
DoGSiteScorer
Comparison to KLIFS pocket
Contents in Practical¶
Binding site detection using DoGSiteScorer
Job submission of structure of interest
Get DoGSiteScorer pocket metadata
Pick the most suitable pocket
Get best binding site file content
Investigate detected pocket
Comparison between DoGSiteScorer and KLIFS pocket
Get DoGSiteScorer pocket residues
Get KLIFS pocket residues
Overlap of pocket residues
References¶
Prediction, Analysis, and Comparison of Active Sites Volkamer et al., (2018), book chapter in Applied Chemoinformatics: Achievements and Future Opportunities, Wiley
DoGSiteScorer, Volkamer et al., J.Chem.Inf.Model, (2012), 52(2):360-372
ProteinsPlus: a web portal for structure analysis of macromolecules. Fährrolfes et al., NAR, (2017), 3;45(W1)
KLIFS: a structural kinase-ligand interaction database, Kanev et al., NAR, (2021), 49(D1):D562-D569
[1]:
import sys
if "google.colab" in sys.modules:
%pip install teachopencadd --no-deps -q
!teachopencadd -d 14
%pip uninstall teachopencadd -y -q
%pip install -qr requirements.txt
Theory¶
Protein binding sites¶
Most biological processes are guided through (non-)reversible binding of molecules. Given a therapeutic target associated to a specific disease, knowing its binding site(s), i.e. the key to the proteins function, is of utmost importance for designing new drugs.
Depending on the given data, e.g., no protein-ligand complex structure (x-ray) is available or one is interested in allosteric sites, binding site detection algorithms come into play. Binding sites, or in the case of enzymes rather called active sites, are cavities in 3-dimensional space, mostly on the surface of a protein structure, that serve as binding (docking) regions for ligands, peptides, or proteins. To interact with each other, the two binding partners need to be complementary concerning shape and physico-chemical properties (key and look principle).
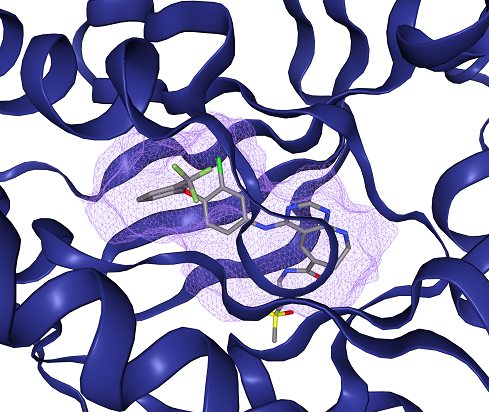
Figure 1: Example of a detected binding site for EGFR kinase (PDB: 3w32) using DoGSiteScorer from proteins.plus. Protein shown as blue cartoon, ligand as sticks (carbons in gray) and binding site as violet cloud (largest subpocket SP_0_0 shown).
Binding site detection¶
Methods overview¶
If ligand information is available, i.e., a protein-ligand complex, the protein residues surrounding the ligand can simply be defined as pocket (e.g. using all protein residues within a predefined radius of the ligand atoms such as 6 Å). If the ligand is absent, prediction tools can be used for in silico pocket detection. These methods can be grouped on the one hand in geometry- and energy-based methods as well as on the other hand in grid-based and grid-free approaches, as outlined in Figure 2. Note that in recent years, more and more machine or deep learning based methods have been developed (see e.g. DeepSite by Jiménez et al., Bioinformatics, 2017, 33(19), 3036–3042).
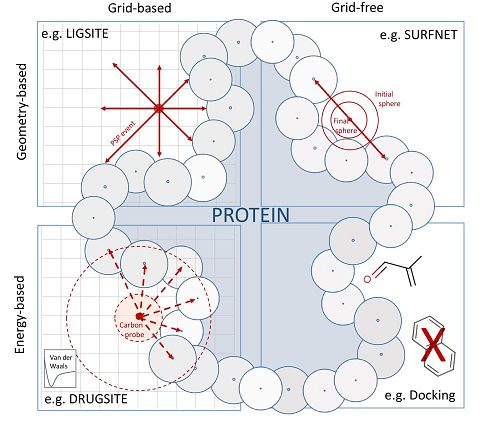
Figure 2: Binding site detection methods can be grouped into geometry-based and energy-based approaches as well as grid-based and grid-free approaches. Figure taken from Prediction, Analysis, and Comparison of Active Sites, Volkamer et al., (2018), Applied Chemoinformatics: Achievements and Future Opportunities, Wiley.
Geometry-based approaches analyze the shape of a molecular surface to locate cavities and incorporate the 3D spatial arrangement of the atoms on the protein surface. Energy-based approaches record interactions of probes or molecular fragments with the protein, thus, favorable energetic responses are assigned to pockets. Both strategies can be performed on a Cartesian grid-based representation of the protein (i.e. checking the environment per grid point) or without (i.e. grid-free).
In the following, an example for each of the four categories will be shortly introduced:
Geometric, grid-based approach: In LIGSITE (Hendlich, et al., J Mol Graph Model., 1997, 15(6):359-63, 389), a Cartesian grid (e.g. 1Å grid spacing) is spanned over the protein of interest. Each grid point is then scanned in seven direction (along the x, y and z axes as well as the four cubic diagonals) and the number of Protein-Solvent-Protein (PSP) events per point is stored (# rays restricted on both side by the protein). Finally, grid points that are buried (= have a high PSP value) are clustered to pockets.
Geometric, grid-free approach: In SURFNET (Laskowski, J Mol Graph., 1995, 13(5):323-30, 307-8), spheres are placed midway between all pairs of atoms on the protein surface directly. In case a probe clashes with any nearby atom, its radius is reduced until no overlap occurs. The resulting probes define the cavities.
Energy, grid-based approach: In DrugSite (An, et al., Genome Informatics, 2004, 15(2): 31–41), the protein is embedded in a Cartesian grid and carbon probes are placed on each grid point. Then, van der Waals energies between the probe and the protein environment within 8 Å distance are calculated. Grid points with unfavorable energies, i.e., above an energy cut-off based on the mean energy and standard deviation over the whole grid, are discarded. Finally, grid points fulfilling this cut-off are merged to pockets.
Energy, grid-free approach: In docking-based methods, fragments (or small molecules) are docked against the protein of interest (placed and scored, for more info on docking see talktorial T015). Pockets are then assigned based on the quantity of fragments that bind to a specific area.
DoGSiteScorer¶
In this talktorial, we will use the DoGSiteScorer functionality, available within protein.plus, to detect and score the pockets of a protein of interest. Thus, the algorithm will be explained in a bit more detail (see Figure 3 for a visual explanation).
Pocket detection: DoGSiteScorer incorporates a geometric and grid-based algorithm to detect pockets. The protein is embedded in a Cartesian grid, and each grid point is labeled as either 0 (free) or 1 (occupied), depending on if it lies within any protein atom’s van der Waals radius. Then, an edge-detection algorithm from image processing, a Difference of Gaussian filter (thus the name DoGSite) is invoked to identify protrusion on the protein surface (i.e. the positions on a protein surface where the location of a sphere-like object is favorable). Based on specific cut-off criteria, grid points with the highest intensity are selected and first clustered to subpockets, then merged to pockets.
- Descriptor calculation: Based on the grid representation of the respective pocket as well as the surrounding protein atoms, properties describing the pocket are derived. These include properties such as volume, surface, or depth of the pocket (calculated directly from the properties of the individual grid points) as well as hyprophobicity, number of available hydrogen bond donors/acceptors or amino acid count (derived from the neighboring protein residues).
Druggability estimates: Additionally, the tool has an in-built druggability predictor. Druggability can be defined as the ability of a (disease-associated) target to bind - and potentially be modulated by - low molecular weight compounds (sometimes also referred to as ligandability). In DoGSiteScorer, druggablity is predicted using a support vector machine (SVM) model, trained and tested on the freely available (non-redundant) druggability data set (NR) DD. The DD consists of 1069 targets and each target was assigned to one of the three classes: druggable, difficult, or undruggable.
For more detail on DoGSiteScorer’s druggability model, see Volkamer et al., J. Chem. Inf. Model., 2012, 52, 2, 360–372. For more info on the druggability concept itself refer to e.g. Hopkins and Groom, The druggable genome. Nat Rev Drug Discov 1, 727–730 (2002).
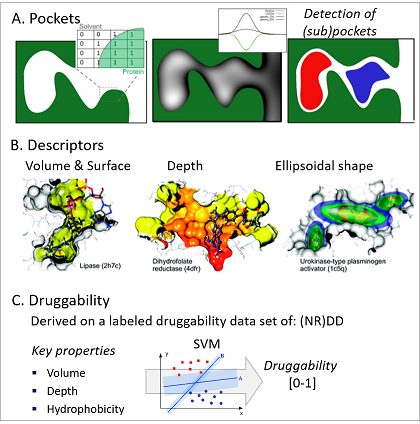
Figure 3: Schematic representation of the individual steps within DoGSiteScorer: A. Pocket detection, B. descriptor calculation and C. druggability estimation. Figure newly composed based on Volkamer et al., J. Chem. Inf. Model., 2012, 52(2), 360–372 and Volkamer et al., J. Chem. Inf. Model., 2010, 50(11), 2041–2052.
Comparison to KLIFS pocket¶
Once we obtain the binding site of interest from DoGSiteScorer, we can compare the results with any other method in order to validate it. Here, we compare it with the KLIFS binding pocket definition for our target kinase structure using the KLIFS API (see Talktorial T012 for more detail).
KLIFS pocket definition in a nutshell: The KLIFS (Kinase-Ligand Interaction Fingerprints and Structures) database is a structural repository of information on over 3600 human and mouse kinase structures. The curated KLIFS data allows systematic analyses of all kinase structures and binding sites, bound ligands and protein-ligand interactions. KLIFS comes with a nomenclature of typical structural motifs within kinases (such as DFG-in/out, hinge region, …) and maps the binding site of all known kinases to 85 residues, defined via an elaborated multiple sequence alignment. It is possible to compare the interaction patterns of kinase-inhibitors to each other to, for example, identify crucial interactions determining kinase-inhibitor selectivity.
In this talktorial, we will query the KLIFS API to return the binding pocket of a specific protein kinase structure of interest for further analysis.
Practical¶
In this practical part, we will introduce how to query the proteins.plus server for binding site detection using DoGSiteScorer for our protein of interest.
Import all necessary libraries.
[2]:
import io
from pathlib import Path
import time
import gzip
import requests
from matplotlib_venn import venn2
import matplotlib.pyplot as plt
import pandas as pd
from biopandas.pdb import PandasPdb
import pypdb
import nglview
from opencadd.databases.klifs import setup_remote
pd.set_option("display.max_columns", 50)
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/swagger_spec_validator/validator12.py:18: DeprecationWarning: jsonschema.RefResolver is deprecated as of v4.18.0, in favor of the https://github.com/python-jsonschema/referencing library, which provides more compliant referencing behavior as well as more flexible APIs for customization. A future release will remove RefResolver. Please file a feature request (on referencing) if you are missing an API for the kind of customization you need.
from jsonschema import RefResolver
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/swagger_spec_validator/validator12.py:18: DeprecationWarning: jsonschema.RefResolver is deprecated as of v4.18.0, in favor of the https://github.com/python-jsonschema/referencing library, which provides more compliant referencing behavior as well as more flexible APIs for customization. A future release will remove RefResolver. Please file a feature request (on referencing) if you are missing an API for the kind of customization you need.
from jsonschema import RefResolver
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/swagger_spec_validator/ref_validators.py:14: DeprecationWarning: jsonschema.RefResolver is deprecated as of v4.18.0, in favor of the https://github.com/python-jsonschema/referencing library, which provides more compliant referencing behavior as well as more flexible APIs for customization. A future release will remove RefResolver. Please file a feature request (on referencing) if you are missing an API for the kind of customization you need.
from jsonschema.validators import RefResolver
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/swagger_spec_validator/validator20.py:18: DeprecationWarning: jsonschema.RefResolver is deprecated as of v4.18.0, in favor of the https://github.com/python-jsonschema/referencing library, which provides more compliant referencing behavior as well as more flexible APIs for customization. A future release will remove RefResolver. Please file a feature request (on referencing) if you are missing an API for the kind of customization you need.
from jsonschema.validators import RefResolver
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/swagger20_validator.py:6: DeprecationWarning: jsonschema.RefResolver is deprecated as of v4.18.0, in favor of the https://github.com/python-jsonschema/referencing library, which provides more compliant referencing behavior as well as more flexible APIs for customization. A future release will remove RefResolver. Please file a feature request (on referencing) if you are missing an API for the kind of customization you need.
from jsonschema import RefResolver
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/swagger20_validator.py:6: DeprecationWarning: jsonschema.RefResolver is deprecated as of v4.18.0, in favor of the https://github.com/python-jsonschema/referencing library, which provides more compliant referencing behavior as well as more flexible APIs for customization. A future release will remove RefResolver. Please file a feature request (on referencing) if you are missing an API for the kind of customization you need.
from jsonschema import RefResolver
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/spec.py:14: DeprecationWarning: jsonschema.RefResolver is deprecated as of v4.18.0, in favor of the https://github.com/python-jsonschema/referencing library, which provides more compliant referencing behavior as well as more flexible APIs for customization. A future release will remove RefResolver. Please file a feature request (on referencing) if you are missing an API for the kind of customization you need.
from jsonschema.validators import RefResolver
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/bravado_core/model.py:888: DeprecationWarning: jsonschema.RefResolver.in_scope is deprecated and will be removed in a future release.
with spec.resolver.in_scope(additional_uri):
Add globals to this talktorial’s path (HERE) and its data folder (DATA).
[3]:
HERE = Path(_dh[-1])
DATA = HERE / "data"
DATATMP = DATA / "_tmp"
DATATMP.mkdir(parents=True, exist_ok=True)
Binding site detection using DoGSiteScorer¶
We first define a function to query the server for a protein of interest. Infos on the REST DoGSite3 API can be found here.
[4]:
def submit_dogsite3_job_with_pdbid(pdb_code, chain_id, ligand=""):
"""
Submit a PDB ID to ProteinsPlus DoGSite3 API and get back the job URL.
Parameters
----------
pdb_code : str
4-letter valid PDB ID, e.g. '3w32'.
chain_id : str
Chain ID, e.g. 'A'.
ligand : str
Name of ligand bound to PDB structure with pdb_id, e.g. 'W32_A_1101'.
Returns
-------
str
The job location URL (relative ID) to poll for results.
"""
url = "https://proteins.plus/api/dogsite3_rest"
payload = {
"dogsite3": {
"pdbCode": pdb_code,
"analysisDetail": "1", # include subpockets
"bindingSitePredictionGranularity": "1", # include druggability scores
"ligand": ligand,
"chain": chain_id,
"ligandBias": "0", # default: no ligand bias
}
}
headers = {"Content-Type": "application/json", "Accept": "application/json"}
r = requests.post(url, json=payload, headers=headers)
r.raise_for_status()
resp = r.json()
# Should return status_code + location (job ID)
if "location" in resp:
return resp["location"]
else:
raise RuntimeError("Unexpected response from DoGSite3 submission: %r" % resp)
Job submission for structure of interest¶
Define the protein of interest. As an example, we chose an EGFR kinase structure, more details can be found in the respective PDB entry: 3w32.
[5]:
pdb_id = "3w32"
chain_id = "A"
# Ligand id manually looked-up from DoGSiteScorer within proteins.plus
# Note that it is generally composed of [pdb_lig_id]_[chain_id]_[pdb_residue_id]
ligand_id = "W32_A_1101"
Submit the query and identify the job location (URL) on the web server for further investigation.
[6]:
job_location = submit_dogsite3_job_with_pdbid(pdb_id, chain_id, ligand_id)
job_location
[6]:
'https://proteins.plus/api/dogsite3_rest/f4AQNSrkroLhcSk11p94Zfh5'
Get DoGSiteScorer pocket metadata¶
We now define a function that collects all data returned by the server and stores them in a pandas DataFrame.
[7]:
def get_dogsitescorer_metadata(job_location, attempts=30):
"""
Get results from a DoGSiteScorer query, i.e., the binding sites which are found over the protein surface,
in the form of a table with the details about all detected pockets.
Parameters
----------
job_location : str
Consists of the location of a finished DoGSiteScorer job on the proteins.plus web server.
attempts : int
The time waiting for the feedback from DoGSiteScorer service.
Returns
-------
pandas.DataFrame
Table with metadata on detected binding sites.
"""
print(f"Querying for job at URL {job_location}...", end="")
while attempts:
# Get job results
result = requests.get(job_location)
result.raise_for_status()
# Get URL of result table file
response = result.json()
if "result_table" in response:
result_file = response["result_table"]
break
attempts -= 1
print(".", end="")
time.sleep(10)
# Get result table (as string)
result_table = requests.get(result_file).text
# Load the table (csv format using "\t" as separator) with pandas DataFrame
# We cannot load the table from a string directly but from a file
# Use io.StringIO to wrap this string as file-like object as needed for read_csv method
# See more: https://docs.python.org/3/library/io.html#io.StringIO
result_table_df = pd.read_csv(io.StringIO(result_table), sep="\t").set_index("name")
return result_table_df
All detected pockets from DoGSiteScorer web server are retrieved and a dataframe is returned containing all pocket descriptor information.
[8]:
metadata = get_dogsitescorer_metadata(job_location)
metadata.head()
Querying for job at URL https://proteins.plus/api/dogsite3_rest/f4AQNSrkroLhcSk11p94Zfh5...
[8]:
| lig_cov | poc_cov | lig_name | 4A_crit | ligSASRatio | volume | enclosure | surface | lipoSurface | depth | surf/vol | lid/hull | ellVol | ell_c/a | ell_b/a | surfGPs | lidGPs | hullGPs | siteAtms | accept | donor | posAtms | negAtms | aromat | hydroAtms | ... | GLY | HIS | ILE | LEU | LYS | MET | PHE | PRO | SER | THR | TRP | TYR | VAL | A | C | G | U | I | N | DA | DC | DG | DT | DN | UNK | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| name | |||||||||||||||||||||||||||||||||||||||||||||||||||
| P_1 | 89.74360 | 99.66560 | W32_A_1101 | 1 | 0.121808 | 306.176 | 0.938650 | 376.060 | 244.070 | 15.71750 | 1.22825 | 0.061350 | 377.02800 | 0.081347 | 0.116841 | 459 | 30 | 489 | 61 | 10 | 10 | 1 | 0 | 5 | 34 | ... | 1 | 0 | 2 | 7 | 1 | 2 | 2 | 0 | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| P_2 | 0.00000 | 0.00000 | none | 0 | 0.000000 | 151.552 | 0.976654 | 378.645 | 212.224 | 9.76524 | 2.49845 | 0.023346 | 84.03600 | 0.202921 | 0.278810 | 251 | 6 | 257 | 41 | 9 | 5 | 0 | 0 | 0 | 20 | ... | 0 | 0 | 1 | 4 | 1 | 0 | 0 | 1 | 2 | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| P_3 | 0.00000 | 0.00000 | none | 0 | 0.000000 | 84.992 | 0.923077 | 244.361 | 151.512 | 6.54828 | 2.87510 | 0.076923 | 16.12450 | 0.386256 | 0.481409 | 132 | 11 | 143 | 27 | 6 | 3 | 1 | 1 | 0 | 18 | ... | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| P_4 | 0.00000 | 0.00000 | none | 0 | 0.000000 | 76.800 | 0.875000 | 289.430 | 200.108 | 6.14492 | 3.76863 | 0.125000 | 12.47730 | 0.460315 | 0.577608 | 112 | 16 | 128 | 26 | 7 | 1 | 0 | 2 | 1 | 14 | ... | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| P_5 | 7.69231 | 3.30579 | W32_A_1101 | 1 | 0.121808 | 61.952 | 0.828829 | 258.652 | 145.068 | 6.64530 | 4.17504 | 0.171171 | 8.06223 | 0.302690 | 0.332725 | 92 | 19 | 111 | 26 | 6 | 5 | 2 | 1 | 1 | 10 | ... | 2 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
5 rows × 69 columns
[9]:
metadata.columns
[9]:
Index(['lig_cov', 'poc_cov', 'lig_name', '4A_crit', 'ligSASRatio', 'volume',
'enclosure', 'surface', 'lipoSurface', 'depth', 'surf/vol', 'lid/hull',
'ellVol', 'ell_c/a', 'ell_b/a', 'surfGPs', 'lidGPs', 'hullGPs',
'siteAtms', 'accept', 'donor', 'posAtms', 'negAtms', 'aromat',
'hydroAtms', 'hydrophobicity', 'metal', 'Cs', 'Ns', 'Os', 'Ss', 'Xs',
'acidicAA', 'basicAA', 'polarAA', 'apolarAA', 'sumAA', 'ALA', 'ARG',
'ASN', 'ASP', 'CYS', 'GLN', 'GLU', 'GLY', 'HIS', 'ILE', 'LEU', 'LYS',
'MET', 'PHE', 'PRO', 'SER', 'THR', 'TRP', 'TYR', 'VAL', 'A', 'C', 'G',
'U', 'I', 'N', 'DA', 'DC', 'DG', 'DT', 'DN', 'UNK'],
dtype='object')
Pick the most suitable pocket¶
The pockets are initially sorted by the volume of the respective pocket. We want to have a deeper look at the pocket that contains the ligand (see ligand coverage column lig_cov) and which is covered by the ligand the most (pocket coverage poc_cov).
For clarity, we drop a few columns first from the dataframe.
[10]:
metadata = metadata[
[
"lig_cov",
"poc_cov",
"lig_name",
"volume",
"enclosure",
"surface",
"depth",
"surf/vol",
"accept",
"donor",
"hydrophobicity",
"metal",
]
]
Sort the obtained binding site by your column of interest.
[11]:
metadata.sort_values(by=["lig_cov", "poc_cov"], ascending=False).head()
# NBVAL_CHECK_OUTPUT
[11]:
| lig_cov | poc_cov | lig_name | volume | enclosure | surface | depth | surf/vol | accept | donor | hydrophobicity | metal | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| name | ||||||||||||
| P_1 | 89.74360 | 99.66560 | W32_A_1101 | 306.176 | 0.938650 | 376.060 | 15.71750 | 1.22825 | 10 | 10 | 0.704918 | 0 |
| P_5 | 7.69231 | 3.30579 | W32_A_1101 | 61.952 | 0.828829 | 258.652 | 6.64530 | 4.17504 | 6 | 5 | 0.576923 | 0 |
| P_2 | 0.00000 | 0.00000 | none | 151.552 | 0.976654 | 378.645 | 9.76524 | 2.49845 | 9 | 5 | 0.658537 | 0 |
| P_3 | 0.00000 | 0.00000 | none | 84.992 | 0.923077 | 244.361 | 6.54828 | 2.87510 | 6 | 3 | 0.666667 | 0 |
| P_4 | 0.00000 | 0.00000 | none | 76.800 | 0.875000 | 289.430 | 6.14492 | 3.76863 | 7 | 1 | 0.692308 | 0 |
Note: We decided here to select the pocket with the best ligand and pocket coverage to have a precise description of the pocket for our comparison later. In other scenarios, you might want to simply sort by hydrophobicity (metadata.sort_values(by='hydrophobicity', ascending=False)) or other values, such as volume.
We define a function that returns the ID (variable is called best_pocket_id) of the pocket that fullfills our sorting criterion.
[12]:
def select_best_pocket(metadata, sorted_by):
"""
This function uses the defined sorting parameter to identify
the best pocket among the obtained pockets.
Parameters
----------
metadata : pd.DataFrame
Pockets retrieved from the DoGSiteScorer website
by : str
Method name(s) to sort table.
Returns
-------
str
Best binding site name.
"""
by_methods = ["volume", "lig_cov", "poc_cov"]
# Sort by the selected method
if all(elem in by_methods for elem in sorted_by):
sorted_pocket = metadata.sort_values(sorted_by, ascending=False)
else:
raise ValueError(f'Selection method not in list: {", ".join(by_methods)}')
# Get name of best pocket
best_pocket_name = sorted_pocket.iloc[0, :].name
return best_pocket_name
We now programatically access the name of the best pocket based on the selected sorting.
[13]:
best_pocket_id = select_best_pocket(metadata, ["lig_cov", "poc_cov"])
best_pocket_id
# NBVAL_CHECK_OUTPUT
[13]:
'P_1'
Get binding site file content¶
We define two helper functions to first get the URL for all pocket pdb or ccp4 files, respectively. Then, we only process the files further for our pocket of interest.
Note that DoGSiteScorer returns two structural (3D) file formats per pocket:
pdb: These files contain all atoms/residues surrounding the pocket.
Accessed via
residuesNaming scheme, e.g. 3w32_P_1
_res.pdb
ccp4: These files contain the grid points composing the pocket, also considered the volume of the pocket.
Accessed via
pocketsNaming scheme, e.g. 3w32_P_1
_gps.ccp4.gz
[14]:
def get_url_for_pockets(job_location, file_type="pdb"):
"""
Get all pocket file locations for a finished DoGSiteScorer job
for a selected file type (pdb/ccp4).
Parameters
----------
job_location : str
URL of finished job submitted to the DoGSiteScorer web server.
file_type : str
Type of file to be returned (pdb/ccp4).
Returns
-------
list
List of all respective pocket file URLs.
"""
# Get job results
result = requests.get(job_location)
if file_type == "pdb":
# Get pocket residues
return result.json()["residues"]
elif file_type == "ccp4":
# Get pocket volumes
return result.json()["pockets"]
else:
raise ValueError(f"File type {file_type} not available.")
[15]:
def get_selected_pocket_location(job_location, best_pocket, file_type="pdb"):
"""
Get the selected binding site file location.
Parameters
----------
job_location : str
URL of finished job submitted to the DoGSiteScorer web server.
best_pocket : str
Selected pocket id.
file_type : str
Type of file to be returned (pdb/ccp4).
Returns
------
str
URL of selected pocket file on the DoGSiteScorer web server.
"""
result = []
# Get URL for all available pdb or ccp4 files
pocket_files = get_url_for_pockets(job_location, file_type)
for pocket_file in pocket_files:
if file_type == "pdb":
if f"{best_pocket}_res" in pocket_file:
result.append(pocket_file)
elif file_type == "ccp4":
if f"{best_pocket}_gps" in pocket_file: # previusly _gpsAll
result.append(pocket_file)
if len(result) > 1:
raise TypeError(f'Multiple strings detected: {", ".join(result)}.')
elif len(result) == 0:
raise TypeError(f"No string detected.")
else:
pass
return result[0]
Get URL for PDB file containing the selected pocket’s residues.
[16]:
selected_pocket_residues_url = get_selected_pocket_location(job_location, best_pocket_id, "pdb")
selected_pocket_residues_url
[16]:
'https://proteins.plus/results/dogsite3/f4AQNSrkroLhcSk11p94Zfh5/3w32_P_1_res.pdb'
Investigate detected pocket¶
Pocket descriptors
The algorithm returns information on all calculated descriptors for the respective pocket.
[17]:
metadata.loc[best_pocket_id]
# NBVAL_CHECK_OUTPUT
[17]:
lig_cov 89.7436
poc_cov 99.6656
lig_name W32_A_1101
volume 306.176
enclosure 0.93865
surface 376.06
depth 15.7175
surf/vol 1.22825
accept 10
donor 10
hydrophobicity 0.704918
metal 0
Name: P_1, dtype: object
Visualize the pocket
We need to get the respective ccp4 file that contains the pocket information. First, we get the URL for theccp4 file containing the selected pocket’s volume.
[18]:
selected_pocket_volume_url = get_selected_pocket_location(job_location, best_pocket_id, "ccp4")
selected_pocket_volume_url
[18]:
'https://proteins.plus/results/dogsite3/f4AQNSrkroLhcSk11p94Zfh5/3w32_P_1_gps.ccp4.gz'
We get the grid point coordinates from that proteins.plus URL. We do not save this to disk, but to a memory file (io.BytesIO). Thanks to the extension in the URL, we know this is a gzipped (.gz extension) CCP4 map (.ccp4). What we do:
Download the file from the server into memory. At this point we have gzipped bytes.
We decompress the response in memory using
gzip.decompress.We wrap it in a
io.BytesIOobject that will behave as an opened file.
[19]:
r = requests.get(selected_pocket_volume_url)
r.raise_for_status()
# Decompress response content and wrap it in a file-like object
memfile = io.BytesIO(gzip.decompress(r.content))
Note: If the pocket volume does not show in the NGLviewer, please try to run the notebook with Jupyter Notebook (instead of Jupyter Lab).
[20]:
def get_pdb_nglviewer(pdb_id):
# download the PDB file content.
pdb_file_content = pypdb.get_pdb_file(pdb_id, filetype="pdb")
# create an nglview structure object from the content
structure = nglview.TextStructure(pdb_file_content, ext="pdb")
# set structure viewer
nglviewer = nglview.show_file(structure)
return nglviewer
[21]:
# viewer = nglview.show_pdbid(pdb_id)
viewer = get_pdb_nglviewer(pdb_id)
# Since we are passing bytes with no path or extension
# we need to tell nglview what kind of data this is: ccp4
viewer.add_component(memfile, ext="ccp4")
viewer.center()
viewer
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/pypdb/pypdb.py:497: DeprecationWarning: The `get_pdb_file` function within pypdb.py is deprecated.See `pypdb/clients/pdb/pdb_client.py` for a near-identical function to use
warnings.warn(
Sending GET request to https://files.rcsb.org/download/3w32.pdb to fetch 3w32's pdb file as a string.
[22]:
viewer.render_image(trim=True, factor=2)
viewer._display_image()
[22]:

The above rendered image shows the same content as when using the proteins.plus web server directly (see Figure 1).
Comparison between DoGSiteScorer and KLIFS pocket¶
Get DoGSiteScorer pocket residues¶
Since we want to compare the automatically predicted pocket with the manually assigned KLIFS pocket, we need to identify the pocket residues.
[23]:
def get_pocket_residues(pocket_url):
"""
Gets residue IDs and names of a specified pocket (via URL).
Parameters
----------
pocket_url : str
URL of selected pocket file on the DoGSiteScorer web server.
Returns
-------
pandas.DataFrame
Table of residues names and IDs for the selected binding site.
"""
# Retrieve PDB file content from URL
result = requests.get(pocket_url)
# Get content of PDB file
pdb_residues = result.text
# Load PDB format as DataFrame
ppdb = PandasPdb().read_pdb_from_list(pdb_residues.splitlines(True))
pdb_df = ppdb.df["ATOM"]
# Drop duplicates
# PDB file contains per atom entries, we only need per residue info
pdb_df.sort_values("residue_number", inplace=True)
pdb_df.drop_duplicates(subset="residue_number", keep="first", inplace=True)
return pdb_df[["residue_number", "residue_name"]]
We get the residues of selected pocket and show the top 5 residues.
[24]:
dogsite_pocket_residues_df = get_pocket_residues(selected_pocket_residues_url)
dogsite_pocket_residues_df.head()
[24]:
| residue_number | residue_name | |
|---|---|---|
| 0 | 718 | LEU |
| 8 | 726 | VAL |
| 15 | 743 | ALA |
| 20 | 744 | ILE |
| 28 | 745 | LYS |
Another pocket visualization
We can also use the selected pocket residues to investigate our pocket in 3D.
[25]:
# Show protein structure as cartoon
viewer = get_pdb_nglviewer(pdb_id)
# Select pocket residues
selection = (
f":{chain_id} and ({' or '.join(dogsite_pocket_residues_df['residue_number'].astype(str))})"
)
# Show pocket residues as stick
viewer.add_representation("ball+stick", selection=selection, aspectRatio=1.0)
# Show ligand as ball and stick
viewer.add_representation("ball+stick", selection=ligand_id.split("_")[0])
viewer.center()
viewer
Sending GET request to https://files.rcsb.org/download/3w32.pdb to fetch 3w32's pdb file as a string.
/opt/miniconda3/envs/T014_3110/lib/python3.11/site-packages/pypdb/pypdb.py:497: DeprecationWarning: The `get_pdb_file` function within pypdb.py is deprecated.See `pypdb/clients/pdb/pdb_client.py` for a near-identical function to use
warnings.warn(
[26]:
viewer.render_image(trim=True, factor=2)
viewer._display_image()
[26]:
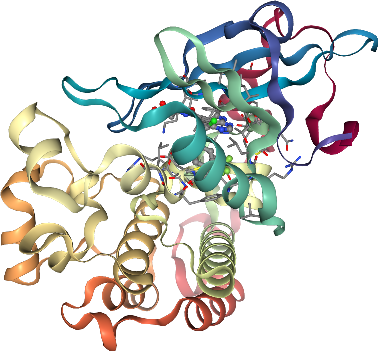
Get KLIFS pocket residues¶
We are using the opencadd.databases.klifs module to extract the binding site residues (PDB residue numbering) as defined in the KLIFS database.
Please refer to Talktorial T012 for detailed information about KLIFS and the opencadd.databases.klifs module usage.
[27]:
session = setup_remote()
[28]:
# Get first structure KLIFS ID associated with PDB ID
structures = session.structures.by_structure_pdb_id(pdb_id)
structure_klifs_id = structures["structure.klifs_id"].iloc[0]
[29]:
# Get the structure's pocket
klifs_pocket_df = session.pockets.by_structure_klifs_id(structure_klifs_id)
# Residue ID: Cast string to int
klifs_pocket_df = klifs_pocket_df.astype({"residue.id": int})
klifs_pocket_df.head()
# NBVAL_CHECK_OUTPUT
[29]:
| residue.klifs_id | residue.id | residue.klifs_region_id | residue.klifs_region | residue.klifs_color | |
|---|---|---|---|---|---|
| 0 | 1 | 716 | I.1 | I | khaki |
| 1 | 2 | 717 | I.2 | I | khaki |
| 2 | 3 | 718 | I.3 | I | khaki |
| 3 | 4 | 719 | g.l.4 | g.l | green |
| 4 | 5 | 720 | g.l.5 | g.l | green |
Overlap of pocket residues¶
[30]:
# Get DoGSiteScorer pocket residues as list
dogsite_pocket_residues = dogsite_pocket_residues_df["residue_number"].to_list()
print(
f"DoGSiteScorer pocket {best_pocket_id} with {len(dogsite_pocket_residues)} residues detected."
)
# Get KLIFS pocket residues as list
klifs_pocket_residues = klifs_pocket_df["residue.id"].to_list()
print(f"KLIFS pocket with {len(klifs_pocket_residues)} residues detected.")
DoGSiteScorer pocket P_1 with 25 residues detected.
KLIFS pocket with 85 residues detected.
[31]:
overlap = set.intersection(set(dogsite_pocket_residues), set(klifs_pocket_residues))
print(f"Overlap between the two residue sets: {len(overlap)}.")
# NBVAL_CHECK_OUTPUT
Overlap between the two residue sets: 23.
Plot the overlap as Venn diagram.
[32]:
print(f"Residue overlap between the two methods using {best_pocket_id}.")
venn2(
[set(dogsite_pocket_residues), set(klifs_pocket_residues)],
set_labels=("DoGSiteScorer", "KLIFS"),
)
plt.show()
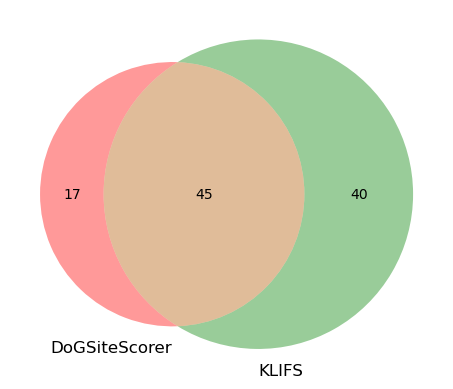
Residue overlap between the two methods using P_1.
As we can see, the subpocket we choose from DoGSiteScorer is very precise. In fact, 23 out of the 25 residues do overlap with the KLIFS definition (85 residues). Though, the KLIFS pocket covers a larger overall pocket area. Out of interest, we can also do the same comparison using the pocket P_2 (instead of pocket P_1) to see how that compares to KLIFS.
[33]:
# Get info for largest DoGSiteScorer pocket P_0
new_pocket_id = "P_2"
dogsite_pocket_residues_df_npi = get_pocket_residues(
get_selected_pocket_location(job_location, new_pocket_id)
)
dogsite_pocket_residues_npi = dogsite_pocket_residues_df_npi["residue_number"].to_list()
print(f"DoGSiteScorer pocket {new_pocket_id} with {len(dogsite_pocket_residues_npi)} detected.")
# NBVAL_CHECK_OUTPUT
DoGSiteScorer pocket P_2 with 16 detected.
[34]:
print(f"Residue overlap between the two methods using {new_pocket_id}.")
venn2(
[set(dogsite_pocket_residues_npi), set(klifs_pocket_residues)],
set_labels=("DoGSiteScorer", "KLIFS"),
)
plt.show()
Residue overlap between the two methods using P_2.
Discussion¶
We introduced the idea of binding site detection algorithms using DoGSiteScorer, which can be queried through the proteins.plus server. Besides investigating into the main (known) binding site, as exemplified above, one can use other predicted pockets to initiate drug design campaigns against a novel, e.g. allosteric or less explored, pocket.
Potential shortcomings: Such algorithms detect the pockets based on a given protein structure (x-ray). If another structure of the same protein is available, applying the algorithm to that structure might lead to (slightly) different pockets due to e.g. conformational changes induced by the bound ligand. Also differences between apo and holo structures do affect the predictions.
Quiz¶
What techniques can be invoked to automatically detect pockets on the protein surface?
What are advantages and disadvantages of automatic binding site detection algorithms?
Try another protein EGFR kinase structure and check the overlap between the predicted residues.