T008 · Protein data acquisition: Protein Data Bank (PDB)¶
Note: This talktorial is a part of TeachOpenCADD, a platform that aims to teach domain-specific skills and to provide pipeline templates as starting points for research projects.
Authors:
Anja Georgi, CADD seminar, 2017, Charité/FU Berlin
Majid Vafadar, CADD seminar, 2018, Charité/FU Berlin
Jaime Rodríguez-Guerra, Volkamer lab, Charité
Dominique Sydow, Volkamer lab, Charité
Talktorial T008: This talktorial is part of the TeachOpenCADD pipeline described in the first TeachOpenCADD publication (J. Cheminform. (2019), 11, 1-7), comprising of talktorials T001-T010.
Aim of this talktorial¶
In this talktorial, we conduct the groundwork for the next talktorial where we will generate a ligand-based ensemble pharmacophore for EGFR. Therefore, we
fetch all PDB IDs for EGFR from the PDB database that fullfil certain criteria (e.g. ligand-bound structures with high resolution),
retrieve protein-ligand structures with the best structural quality,
align all structures, and
extract and save the ligands to be used in the next talktorial.
Contents in Theory¶
Protein Data Bank (PDB)
Query the PDB using the Python packages
biotiteandpypdb
Contents in Practical¶
Select a query protein
Get the number of PDB entries for a query protein
Find PDB entries fullfilling certain conditions
Select PDB entries with the highest resolution
Get metadata of ligands from top structures
Draw top ligand molecules
Create protein-ligand ID pairs
Align PDB structures and extract ligands
References¶
Protein Data Bank (PDB website)
pypdbPython package (Bioinformatics (2016), 1, 159-60; documentation)biotitePython package (BMC Bioinformatics (2018), 19; documentation)Molecular superposition with the Python package
opencadd(repository)
[1]:
import sys
if "google.colab" in sys.modules:
%pip install teachopencadd --no-deps -q
!teachopencadd -d 8
%pip uninstall teachopencadd -y -q
%pip install -qr requirements.txt
Theory¶
Protein Data Bank (PDB)¶
The RCSB Protein Data Bank (PDB) is a comprehensive structural biology information database and a key resource in areas of structural biology, such as structural genomics and drug design (PDB website).
Structural data is generated from structural determination methods such as X-ray crystallography (most common method), nuclear magnetic resonance (NMR), and cryo electron microscopy (cryo-EM). For each entry, the database contains (i) the 3D coordinates of the atoms and the bonds connecting these atoms for proteins, ligand, cofactors, water molecules, and ions, as well as (ii) meta information on the structural data such as the PDB ID, the authors, the deposition date, the structural determination method used, and the structural resolution. The structural resolution is a measure of the collected data quality and has the unit Å (Angstrom); the lower the value, the higher the quality of the structure.
The PDB website offers a 3D visualization of the protein structures (with ligand interactions if available) and a structure quality metrics, as can be seen for the PDB entry of an example epidermal growth factor receptor (EGFR) with the PDB ID 3UG5.
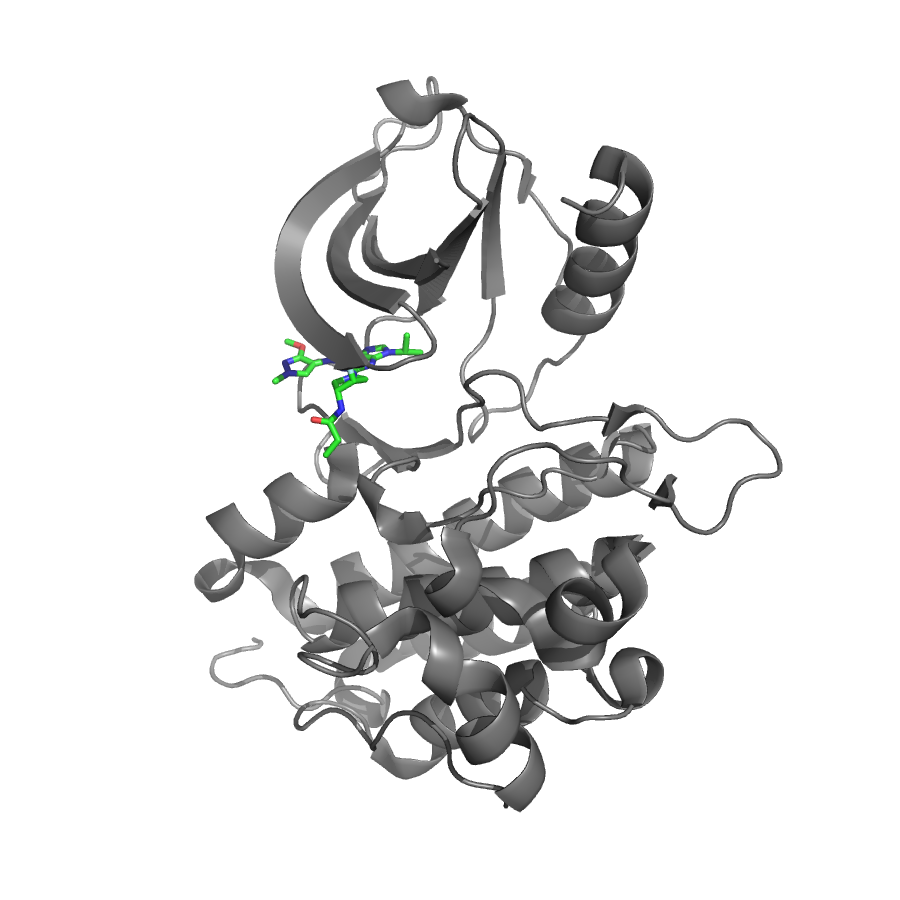
Figure 1: The protein structure (in gray) with an interacting ligand (in green) is shown for an example epidermal growth factor receptor (EGFR) with the PDB ID 3UG5.
Query the PDB using the Python packages biotite and pypdb¶
Each structure in the PDB database is linked to many different fields to hold meta information. Check out the complete list of available fields for chemicals/structures and supported operators on the RCSB website. The Python package biotite provides the convenient module databases.rcsb (see
docs), which allows us to query one (FieldQuery, see docs) or more (CompositeQuery, see docs) of these fields to retrieve a count (count) or list (search) of PDB IDs that
match our criteria.
The Python package pypdb offers an interface for the PDB to not only query for PDB IDs but also to download associated metadata and structural files (Bioinformatics (2016), 1, 159-60, documentation). Check out the demo notebook introducing the pypdb API.
We will use both packages in this notebook: biotite to quickly filter down the many structures in the PDB given certain criteria and pypdb to download metadata and structural files for specific PDB entries of interest.
Practical¶
[2]:
import collections
import logging
import pathlib
import time
import warnings
import datetime
import pandas as pd
import matplotlib.pyplot as plt
from bs4 import BeautifulSoup
import requests
from tqdm.auto import tqdm
import redo
import requests_cache
import nglview
import pypdb
import biotite.database.rcsb as rcsb
from rdkit.Chem import Draw
from rdkit.Chem import PandasTools
from opencadd.structure.superposition.api import align, METHODS
from opencadd.structure.core import Structure
# Disable some unneeded warnings
logger = logging.getLogger("opencadd")
logger.setLevel(logging.ERROR)
warnings.filterwarnings("ignore")
# Cache requests -- this will speed up repeated queries to PDB
requests_cache.install_cache("rcsb_pdb", backend="memory")
[3]:
# Define paths
HERE = pathlib.Path(_dh[-1])
DATA = HERE / "data"
Select a query protein¶
We use EGFR as query protein for this talktorial. The UniProt ID of EGFR is P00533, which will be used in the following to query the PDB database.
[4]:
uniprot_id = "P00533"
Get the number of PDB entries for a query protein¶
How many structures are available in the PDB for EGFR (at the time this notebook was last run)?
[5]:
query_by_uniprot_id = rcsb.FieldQuery(
"rcsb_polymer_entity_container_identifiers.reference_sequence_identifiers.database_accession",
exact_match=uniprot_id,
)
today = datetime.datetime.now()
print(
f"Number of structures on {today.year}-{today.month}-{today.day}: {rcsb.count(query_by_uniprot_id)}"
)
Number of structures on 2026-4-21: 385
How many structures were available in every year since the PDB was established in 1971?
[6]:
# Define lists for years and number of structures available in a given year
years = range(1971, datetime.datetime.now().year)
n_structures = []
for year in years:
# Set latest date for allowed deposition
before_deposition_date = f"{year}-12-31T23:59:59Z"
# Set up query for structures deposited latest at given date
query_by_deposition_date = rcsb.FieldQuery(
"rcsb_accession_info.deposit_date", less_or_equal=before_deposition_date
)
# Set up combined query
query = rcsb.CompositeQuery(
[query_by_uniprot_id, query_by_deposition_date],
"and",
)
# Count matching structures and add to list
n_structures.append(rcsb.count(query))
# wait shortly to not overload the API
time.sleep(0.1)
Plot the results!
[7]:
plt.plot(years, n_structures)
plt.title("PDB entries for EGFR")
plt.xlabel("Year")
plt.ylabel("Number of structures available in a given year");

Find PDB entries fullfilling certain conditions¶
We will search for PDB IDs describing structures in the PDB that fulfill the following criteria:
Structures for UniProt ID P00533: This is our target of interest, EGFR!
Structures deposited before 2020: This step is for TeachOpenCADD-internal maintanence purposes. We will only consider structures that were deposited before 2020. That way the results of this notebook will stay the same over time, allowing us to check with our continuous integration (CI) that this notebook does not go out of service.
Structure resolved by X-ray crystallography: We could include all methods but let’s check out the API on how we can select experimental methods.
Structure has a resolution less than or equal to 3.0: The lower the resolution value, the higher is the quality of the structure, i.e. the certainty that the assigned 3D coordinates of the atoms are correct. Atomic orientations can be determined below 3 Å. Thus, this threshold is often used for structures relevant for structure-based drug design.
Structure has only one chain: We do this to make our lives easier lateron.
Structure has a ligand with molecular weight greater than 100.0 Da: PDB-annotated ligands can be ligands, but also solvents and ions. In order to filter only ligand-bound structures, we keep only structures with annotated ligand of a at least 100.0 Da (many solvents and ions weight less). Note: this is a simple, but not comprehensive exclusion of solvents and ions.
We make use of the biotite package again to query for PDB IDs based on the following combined queries:
Field |
Operator |
Value |
|---|---|---|
|
|
P00533 |
|
|
2020-01-01 |
|
|
X-RAY DIFFRACTION |
|
|
\(3.0\) |
|
|
\(1\) |
|
|
\(100.0\) |
We define our criteria.
[8]:
uniprot_id = "P00533"
before_deposition_date = "2020-01-01T00:00:00Z"
experimental_method = "X-RAY DIFFRACTION"
max_resolution = 3.0
n_chains = 1
min_ligand_molecular_weight = 100.0
We set up each query.
[9]:
query_by_uniprot_id = rcsb.FieldQuery(
"rcsb_polymer_entity_container_identifiers.reference_sequence_identifiers.database_accession",
exact_match=uniprot_id,
)
query_by_deposition_date = rcsb.FieldQuery(
"rcsb_accession_info.deposit_date", less=before_deposition_date
)
query_by_experimental_method = rcsb.FieldQuery("exptl.method", exact_match=experimental_method)
query_by_resolution = rcsb.FieldQuery(
"rcsb_entry_info.resolution_combined", less_or_equal=max_resolution
)
query_by_polymer_count = rcsb.FieldQuery(
"rcsb_entry_info.deposited_polymer_entity_instance_count", equals=n_chains
)
query_by_ligand_mw = rcsb.FieldQuery(
"chem_comp.formula_weight", molecular_definition=True, greater=min_ligand_molecular_weight
)
We perform each of the queries alone and check the number of matches per condition.
[10]:
print(f"Number of structures with UniProt ID {uniprot_id}: {rcsb.count(query_by_uniprot_id)}")
time.sleep(0.1) # wait shortly to not overload the API
print(
f"Number of structures deposited before {before_deposition_date}: {rcsb.count(query_by_deposition_date)}"
)
time.sleep(0.1)
print(
f"Number of structures resolved by {experimental_method}: {rcsb.count(query_by_experimental_method)}"
)
time.sleep(0.1)
print(
f"Number of structures with resolution less than or equal to {max_resolution}: {rcsb.count(query_by_resolution)}"
)
time.sleep(0.1)
print(f"Number of structures with only {n_chains} chain: {rcsb.count(query_by_polymer_count)}")
time.sleep(0.1)
print(
f"Number of structures with ligand of more than or equal to {min_ligand_molecular_weight} Da: {rcsb.count(query_by_ligand_mw)}"
)
Number of structures with UniProt ID P00533: 385
Number of structures deposited before 2020-01-01T00:00:00Z: 166264
Number of structures resolved by X-RAY DIFFRACTION: 203558
Number of structures with resolution less than or equal to 3.0: 201385
Number of structures with only 1 chain: 86880
Number of structures with ligand of more than or equal to 100.0 Da: 252475
We combine all queries with the and operator to match only PDB IDs that fulfill all the conditions.
[11]:
query = rcsb.CompositeQuery(
[
query_by_uniprot_id,
query_by_deposition_date,
query_by_experimental_method,
query_by_resolution,
query_by_polymer_count,
query_by_ligand_mw,
],
"and",
)
pdb_ids = rcsb.search(query)
print(f"Number of matches: {len(pdb_ids)}")
print("Selected PDB IDs:")
print(*pdb_ids)
Number of matches: 110
Selected PDB IDs:
2GS2 2ITN 2ITQ 2ITV 2ITZ 2J5F 3VJO 3W2Q 3W2R 3W32 4I22 4I23 4JQ7 4RJ8 4WKQ 5C8N 5CAO 5EDP 5EDQ 5EM5 5EM6 5HG5 5HG8 5HIC 5U8L 5UG9 5UGC 5X28 5ZTO 5ZWJ 6JWL 6JXT 6S89 6S8A 6S9C 1M17 2ITT 2ITX 3UG1 3UG2 3W2O 3W2P 3W2S 4I1Z 4JR3 4LI5 4LQM 4RJ4 4RJ6 4WRG 4ZAU 5C8K 5CAL 5CAN 5CAP 5CAS 5CAU 5CAV 5EDR 5EM7 5FED 5FEE 5HCY 5X27 5XDL 5XGM 6JRJ 6JRK 6JRX 6JZ0 6S9D 1M14 1XKK 2EB2 2EB3 2ITP 2ITU 2ITW 2RGP 3BEL 3GOP 3POZ 3VJN 3W33 4G5J 4HJO 4JQ8 4JRV 4RJ7 5C8M 5CAQ 5EM8 5GMP 5GNK 5GTZ 5HCX 5HCZ 5HG7 5HG9 5HIB 5J9Y 5J9Z 5UG8 5UGA 5UGB 5X26 5XDK 6D8E 6JX0 6JX4
Select PDB entries with the hightest resolution¶
So far we have used certain search criteria to find PDB entries of interest.
At the moment, we cannot access the structures’ resolution directly via biotite; with biotite we can only check if the resolution fullfils a certain condition. Instead we download the full metadata for our selected PDB IDs. To this end, we use from the pypdb package the method describe_pdb. Each structure’s metadata is returned as a dictionary.
Note: we only fetch meta information on PDB structures here, we do not fetch the structures (3D coordinates), yet.
The
redo.retriableline is a decorator. This wraps the function and provides extra functionality. In this case, it will retry failed queries automatically (10 times maximum).
[12]:
@redo.retriable(attempts=10, sleeptime=2)
def describe_one_pdb_id(pdb_id):
"""Fetch meta information from PDB."""
described = pypdb.describe_pdb(pdb_id)
if described is None:
print(f"! Error while fetching {pdb_id}, retrying ...")
raise ValueError(f"Could not fetch PDB id {pdb_id}")
return described
[13]:
pdbs_data = [describe_one_pdb_id(pdb_id) for pdb_id in tqdm(pdb_ids)]
Let’s take a look at the metadata of the first PDB IDs (keys only because the dictionary contains a lot of information, which we do not want to print here). Find more more information about the PDB metadata in the Beginner’s Guide to PDB Structures and the PDBx/mmCIF Format.
[14]:
print("\n".join(pdbs_data[0].keys()))
audit_author
cell
citation
database_2
diffrn
diffrn_detector
diffrn_radiation
diffrn_source
entry
exptl
exptl_crystal
exptl_crystal_grow
pdbx_audit_revision_category
pdbx_audit_revision_details
pdbx_audit_revision_group
pdbx_audit_revision_history
pdbx_audit_revision_item
pdbx_database_related
pdbx_database_status
pdbx_initial_refinement_model
pdbx_vrpt_summary
pdbx_vrpt_summary_diffraction
pdbx_vrpt_summary_geometry
rcsb_accession_info
rcsb_entry_container_identifiers
rcsb_entry_info
rcsb_primary_citation
refine
refine_hist
refine_ls_restr
reflns
reflns_shell
software
struct
struct_keywords
symmetry
rcsb_id
Let’s take a closer look at two keys that will be of interest to us: The "entry" key which contains the PDB ID ("id") and the "rcsb_entry_info" key which contains amongst others the structure’s resolution ("resolution_combined").
[15]:
pdbs_data[0]["entry"]
[15]:
{'id': '2GS2'}
[16]:
pdbs_data[0]["rcsb_entry_info"]
[16]:
{'assembly_count': 1,
'branched_entity_count': 0,
'cis_peptide_count': 0,
'deposited_atom_count': 2443,
'deposited_deuterated_water_count': 0,
'deposited_hydrogen_atom_count': 0,
'deposited_model_count': 1,
'deposited_modeled_polymer_monomer_count': 305,
'deposited_nonpolymer_entity_instance_count': 0,
'deposited_polymer_entity_instance_count': 1,
'deposited_polymer_monomer_count': 330,
'deposited_solvent_atom_count': 24,
'deposited_unmodeled_polymer_monomer_count': 25,
'diffrn_radiation_wavelength_maximum': 1.116,
'diffrn_radiation_wavelength_minimum': 1.116,
'disulfide_bond_count': 0,
'entity_count': 2,
'experimental_method': 'X-ray',
'experimental_method_count': 1,
'inter_mol_covalent_bond_count': 0,
'inter_mol_metalic_bond_count': 0,
'molecular_weight': 37.56,
'na_polymer_entity_types': 'Other',
'nonpolymer_entity_count': 0,
'polymer_composition': 'homomeric protein',
'polymer_entity_count': 1,
'polymer_entity_count_DNA': 0,
'polymer_entity_count_RNA': 0,
'polymer_entity_count_nucleic_acid': 0,
'polymer_entity_count_nucleic_acid_hybrid': 0,
'polymer_entity_count_protein': 1,
'polymer_entity_taxonomy_count': 1,
'polymer_molecular_weight_maximum': 37.56,
'polymer_molecular_weight_minimum': 37.56,
'polymer_monomer_count_maximum': 330,
'polymer_monomer_count_minimum': 330,
'resolution_combined': [2.8],
'selected_polymer_entity_types': 'Protein (only)',
'software_programs_combined': ['BLU-ICE',
'CNS',
'HKL-2000',
'PDB_EXTRACT',
'PHASER'],
'solvent_entity_count': 1,
'structure_determination_methodology': 'experimental',
'structure_determination_methodology_priority': 10,
'diffrn_resolution_high': {'provenance_source': 'Depositor assigned',
'value': 2.8}}
Now we save the resolution per PDB ID as pandas DataFrame, sorted by the resolution in ascending order.
[17]:
resolution = pd.DataFrame(
[
[pdb_data["entry"]["id"], pdb_data["rcsb_entry_info"]["resolution_combined"][0]]
for pdb_data in pdbs_data
],
columns=["pdb_id", "resolution"],
).sort_values(by="resolution", ignore_index=True)
resolution
[17]:
| pdb_id | resolution | |
|---|---|---|
| 0 | 5UG9 | 1.330 |
| 1 | 5HG8 | 1.420 |
| 2 | 5UG8 | 1.460 |
| 3 | 3POZ | 1.500 |
| 4 | 5HG5 | 1.520 |
| ... | ... | ... |
| 105 | 2ITX | 2.980 |
| 106 | 5GTZ | 2.999 |
| 107 | 2J5F | 3.000 |
| 108 | 5C8K | 3.000 |
| 109 | 4I1Z | 3.000 |
110 rows × 2 columns
Get metadata of ligands from top structures¶
[18]:
top_num = 6 # Number of top structures
In the next talktorial, we will build ligand-based ensemble pharmacophores from the top top_num structures with the highest resolution.
[19]:
selected_pdb_ids = resolution[:top_num]["pdb_id"].to_list()
print(f"Selected PDB IDs: {selected_pdb_ids}")
Selected PDB IDs: ['5UG9', '5HG8', '5UG8', '3POZ', '5HG5', '5UGC']
The selected highest resolution PDB entries can contain ligands targeting different binding sites, e.g. allosteric and orthosteric ligands, which would hamper ligand-based pharmacophore generation. Thus, we will focus on the following 4 structures, which contain ligands in the orthosteric binding pocket. The code provided later in the notebook can be used to verify this.
[20]:
selected_pdb_ids = ["5UG9", "5HG8", "5UG8", "5UGC"]
print(f"Selected PDB IDs (frozen set): {selected_pdb_ids}")
Selected PDB IDs (frozen set): ['5UG9', '5HG8', '5UG8', '5UGC']
We fetch the PDB information about the top top_num ligands using get_ligands, to be stored as csv file (as dictionary per ligand).
If a structure contains several ligands, we select the largest ligand. Note: this is a simple, but not comprehensive method to select a ligand in the binding site of a protein. This approach may also select a cofactor bound to the protein. Therefore, please check the automatically selected top ligands visually before further usage.
[21]:
def get_ligands(pdb_id):
"""
RCSB has not provided a new endpoint for ligand information yet. As a
workaround we are obtaining extra information from ligand-expo.rcsb.org,
using HTML parsing. Check Talktorial T011 for more info on this technique!
"""
pdb_info = _fetch_pdb_nonpolymer_info(pdb_id)
ligand_expo_ids = [
nonpolymer_entities["pdbx_entity_nonpoly"]["comp_id"]
for nonpolymer_entities in pdb_info["data"]["entry"]["nonpolymer_entities"]
]
ligands = {}
for ligand_expo_id in ligand_expo_ids:
ligand_expo_info = _fetch_ligand_expo_info(ligand_expo_id)
ligands[ligand_expo_id] = ligand_expo_info
return ligands
def _fetch_pdb_nonpolymer_info(pdb_id):
"""
Fetch nonpolymer data from rcsb.org.
Thanks @BJWiley233 and Rachel Green for this GraphQL solution.
"""
query = """{
entry(entry_id: "%s") {
nonpolymer_entities {
pdbx_entity_nonpoly {
comp_id
name
rcsb_prd_id
}
}
}
}""" % pdb_id
query_url = f"https://data.rcsb.org/graphql?query={query}"
response = requests.get(query_url)
response.raise_for_status()
info = response.json()
return info
def _fetch_ligand_expo_info(ligand_id):
"""
Fetch ligand data from RCSB API using the exact JSON structure
provided for identifiers like 8AM.
"""
url = f"https://data.rcsb.org/rest/v1/core/chemcomp/{ligand_id.upper()}"
r = requests.get(url)
if r.status_code != 200:
return {}
data = r.json()
chem = data.get("chem_comp", {})
rcsb_desc = data.get("rcsb_chem_comp_descriptor", {})
if isinstance(rcsb_desc, list):
inchi = next(
(item.get("descriptor") for item in rcsb_desc if item.get("type") == "InChI"), None
)
inchikey = next(
(item.get("descriptor") for item in rcsb_desc if item.get("type") == "InChIKey"), None
)
smiles = next(
(item.get("descriptor") for item in rcsb_desc if "SMILES" in item.get("type", "")),
None,
)
else:
inchi = rcsb_desc.get("in_ch_i")
inchikey = rcsb_desc.get("in_ch_ikey")
smiles = rcsb_desc.get("smilesstereo")
if not smiles:
pdbx_descs = data.get("pdbx_chem_comp_descriptor", [])
for d in pdbx_descs:
if d.get("program") == "OpenEye OEToolkits" and d.get("type") == "SMILES_CANONICAL":
smiles = d.get("descriptor")
return {
"Name": chem.get("name"),
"Formula": chem.get("formula"),
"Molecular weight": chem.get("formula_weight"),
"Formal charge": chem.get("pdbx_formal_charge"),
"Atom count": data.get("rcsb_chem_comp_info", {}).get("atom_count"),
"Chiral atom count": data.get("rcsb_chem_comp_info", {}).get("atom_count_chiral"),
"Bond count": data.get("rcsb_chem_comp_info", {}).get("bond_count"),
"Aromatic bond count": data.get("rcsb_chem_comp_info", {}).get("bond_count_aromatic"),
"Component type": chem.get("type"),
# Set keys
"InChI descriptor": inchi,
"InChIKey descriptor": inchikey,
"Stereo SMILES (OpenEye)": smiles,
}
[22]:
columns = [
"@structureId",
"@chemicalID",
"@type",
"@molecularWeight",
"chemicalName",
"formula",
"InChI",
"InChIKey",
"smiles",
]
rows = []
for pdb_id in selected_pdb_ids:
ligands = get_ligands(pdb_id)
# If several ligands contained, take largest (first in results)
ligand_id, properties = max(ligands.items(), key=lambda kv: kv[1]["Molecular weight"])
rows.append(
[
pdb_id,
ligand_id,
properties["Component type"],
properties["Molecular weight"],
properties["Name"],
properties["Formula"],
properties["InChI descriptor"],
properties["InChIKey descriptor"],
properties["Stereo SMILES (OpenEye)"],
]
)
[23]:
# NBVAL_CHECK_OUTPUT
# Change the format to DataFrame
ligands = pd.DataFrame(rows, columns=columns)
ligands
[23]:
| @structureId | @chemicalID | @type | @molecularWeight | chemicalName | formula | InChI | InChIKey | smiles | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 5UG9 | 8AM | non-polymer | 445.494 | N-[(3R,4R)-4-fluoro-1-{6-[(3-methoxy-1-methyl-... | C20 H28 F N9 O2 | None | None | CCC(=O)N[C@@H]1CN(C[C@H]1F)c2nc(c3c(n2)n(cn3)C... |
| 1 | 5HG8 | 634 | non-polymer | 377.400 | N-[3-({2-[(1-methyl-1H-pyrazol-4-yl)amino]-7H-... | C19 H19 N7 O2 | None | None | CCC(=O)Nc1cccc(c1)Oc2c3cc[nH]c3nc(n2)Nc4cnn(c4)C |
| 2 | 5UG8 | 8BP | non-polymer | 415.468 | N-[(3R,4R)-4-fluoro-1-{6-[(1-methyl-1H-pyrazol... | C19 H26 F N9 O | None | None | CCC(=O)N[C@@H]1CN(C[C@H]1F)c2nc(c3c(n2)n(cn3)C... |
| 3 | 5UGC | 8BS | non-polymer | 417.441 | N-[(3R,4R)-4-fluoro-1-{6-[(3-methoxy-1-methyl-... | C18 H24 F N9 O2 | None | None | CCC(=O)N[C@@H]1CN(C[C@H]1F)c2nc(c3c(n2)n(cn3)C... |
[24]:
ligands.to_csv(DATA / "PDB_top_ligands.csv", header=True, index=False)
Draw top ligand molecules¶
[25]:
PandasTools.AddMoleculeColumnToFrame(ligands, "smiles")
Draw.MolsToGridImage(
mols=list(ligands.ROMol),
legends=list(ligands["@chemicalID"] + ", " + ligands["@structureId"]),
molsPerRow=top_num,
)
[25]:

Create protein-ligand ID pairs¶
[26]:
# NBVAL_CHECK_OUTPUT
pairs = collections.OrderedDict(zip(ligands["@structureId"], ligands["@chemicalID"]))
pairs
[26]:
OrderedDict([('5UG9', '8AM'),
('5HG8', '634'),
('5UG8', '8BP'),
('5UGC', '8BS')])
Align PDB structures and extract ligands¶
Since we want to build ligand-based ensemble pharmacophores in the next talktorial, it is necessary to align all structures to each other in 3D.
We will use the Python package opencadd (repository), which includes a 3D superposition subpackage to guide the structural alignment of the proteins. The approach is based on superposition guided by sequence alignment of provided matched residues. There are other methods in the package, but this simple one will be enough for the task at hand.
Get the PDB structure files¶
We now fetch the PDB structure files, i.e. 3D coordinates of the protein, ligand (and if available other atomic or molecular entities such as cofactors, water molecules, and ions) from the PDB using opencadd.structure.superposition.
Available file formats are pdb and cif, which store the 3D coordinations of atoms of the protein (and ligand, cofactors, water molecules, and ions) as well as information on bonds between atoms. Here, we work with pdb files.
[27]:
# Download PDB structures
structures = [Structure.from_pdbid(pdb_id) for pdb_id in pairs]
structures
[27]:
[<Universe with 2664 atoms>,
<Universe with 2716 atoms>,
<Universe with 2632 atoms>,
<Universe with 2542 atoms>]
Extract protein and ligand¶
Extract protein and ligand from the structure in order to remove solvent and other artifacts of crystallography.
[28]:
complexes = [
Structure.from_atomgroup(structure.select_atoms(f"protein or resname {ligand}"))
for structure, ligand in zip(structures, pairs.values())
]
complexes
[28]:
[<Universe with 2330 atoms>,
<Universe with 2491 atoms>,
<Universe with 2319 atoms>,
<Universe with 2320 atoms>]
[29]:
# Write complex to file
for complex_, pdb_id in zip(complexes, pairs.keys()):
complex_.write(DATA / f"{pdb_id}.pdb")
Align proteins¶
Align complexes (based on protein atoms).
[30]:
results = align(complexes, method=METHODS["mda"])
nglview can be used to visualize molecular data within Jupyter notebooks. With the next cell we will visualize out aligned protein-ligand complexes.
[31]:
view = nglview.NGLWidget()
for complex_ in complexes:
view.add_component(complex_.atoms)
view
[32]:
view.render_image(trim=True, factor=2, transparent=True);
[33]:
view._display_image()
[33]:

Extract ligands¶
[34]:
ligands = [
Structure.from_atomgroup(complex_.select_atoms(f"resname {ligand}"))
for complex_, ligand in zip(complexes, pairs.values())
]
ligands
[34]:
[<Universe with 32 atoms>,
<Universe with 28 atoms>,
<Universe with 30 atoms>,
<Universe with 30 atoms>]
[35]:
for ligand, pdb_id in zip(ligands, pairs.keys()):
ligand.write(DATA / f"{pdb_id}_lig.pdb")
We check the existence of all ligand pdb files.
[36]:
ligand_files = []
for file in DATA.glob("*_lig.pdb"):
ligand_files.append(file.name)
ligand_files
[36]:
['5HG8_lig.pdb', '5UG8_lig.pdb', '5UG9_lig.pdb', '5UGC_lig.pdb']
We can also use nglview to depict the co-crystallized ligands alone. As we can see, the selected complexes contain ligands populating the same binding pocket and can thus be used in the next talktorial for ligand-based pharmacophore generation.
[37]:
view = nglview.NGLWidget()
for component_id, ligand in enumerate(ligands):
view.add_component(ligand.atoms)
view.remove_ball_and_stick(component=component_id)
view.add_licorice(component=component_id)
view
[38]:
view.render_image(trim=True, factor=2, transparent=True);
[39]:
view._display_image()
[39]:

Discussion¶
In this talktorial, we learned how to retrieve protein and ligand meta information and structural information from the PDB. We retained only X-ray structures for EGFR and filtered our data by resolution and ligand availability. Ultimately, we aimed for an aligned set of ligands to be used in the next talktorial for the generation of ligand-based ensemble pharmacophores.
In order to enrich information about ligands for pharmacophore modeling, it is advisable to not only filter by PDB structure resolution, but also to check for ligand diversity (see Talktorial T005 on molecule clustering by similarity) and to check for ligand activity (i.e. to include only potent ligands).
On a technical note: We saw that the different bits of information deposited in the PDB (structural metadata, coordinates, ligand metadata) cannot be accessed at the moment from a single Python package but taking together biotite and pypdb - and a bit of web scraping to extract ligand information - we were able to collect all the pieces of information that we needed. Given that the PDB only recently changed completely to a new API (Nov
2020), related open source Python packages might offer more functionalities in the future after having had the time to catch up.
Quiz¶
Summarize the kind of data that the Protein Data Bank contains.
Explain what the resolution of a structure stands for and how and why we filter for it in this talktorial.
Explain what an alignment of structures means and discuss the alignment performed in this talktorial.