T012 · Data acquisition from KLIFS¶
Note: This talktorial is a part of TeachOpenCADD, a platform that aims to teach domain-specific skills and to provide pipeline templates as starting points for research projects.
Authors:
Dominique Sydow, 2019-2020, Volkamer lab, Charité
Jaime Rodríguez-Guerra, 2019-2020, Volkamer lab, Charité
David Schaller, 2020, Volkamer lab, Charité
Aim of this talktorial¶
KLIFS is a database for kinase-ligand interaction fingerprints and structures. In this talktorial, we will use the programmatic access to this database (KLIFS OpenAPI) and the opencadd (GitHub) package to interact with its rich content. First, we will use a query kinase (EGFR) to fetch all available protein structures and explore their bound ligands as well as interaction
fingerprints. Then, we will explore the bioactivity data for the EGFR inhibitor Gefitinib in order to find off-targets. Last but not least, we offer a convenience function that allows to easily explore different kinases.
Contents in Theory¶
Kinases
EGFR and Gefitinib
KLIFS database
KLIFS OpenAPI
opencadd
Contents in Practical¶
Define kinase and ligand of interest: EGFR and Gefitinib
Generate a KLIFS Python client
Explore the KLIFS OpenAPI
Kinase groups
Kinase families
Kinases
Structures
Interaction fingerprints
Structure coordinates
Ligands
Case study: EGFR (using
opencadd)Get all structures for EGFR
Average interaction fingerprint
Select an example EGFR-Gefitinib structure
Show the structure with
nglviewShow all kinase-bound ligands with
rdkitExplore profiling data for Gefitinib
Explore a random kinase in KLIFS
References¶
Introduction to protein kinases and inhibition (Chapter 9 in Med. Chem. Anticancer Drugs (2008), 251-305)
Kinase classification by Manning et al. (Science (2002), 298, 1912-1934)
Kinase-centric computational drug development (Annu. Rep. Med. Chem. (2017), 50, 197-236)
EGFR and Gefitinib
Review on the EGFR family (Pharmacol. Res. (2019), 139, 395-411 and Front. Cell Dev. Biol. (2016), 8)
EGFR kinase details on UniProt
Gefitinib details on PubChem
KLIFS - a kinase-inhibitor interactions database
Main database/website reference (Nucleic Acids Res. (2020))
Introduction of the KLIFS website & database (Nucleic Acids Res. (2016), 44, 6, D365–D371)
Initial KLIFS dataset, binding mode classification, residue numbering (J. Med. Chem. (2014), 57, 2, 249-277)
NGLView, the interactive molecule visualizer (Bioinformatics (2018), 34, 1241–124)
[1]:
import sys
if "google.colab" in sys.modules:
%pip install teachopencadd --no-deps -q
!teachopencadd -d 12
%pip uninstall teachopencadd -y -q
%pip install -qr requirements.txt
Theory¶
Kinases¶
Protein kinases are one of the most important and well-studied drug targets, since they are critical to most aspects of cell life. Their dysregulation causes many diseases such as cancer, inflammation, and autoimmune disorders.
Protein kinases catalyze the phosphorylation of tyrosine, serine, threonine and histidine residues of themselves or other kinases (Med. Chem. Anticancer Drugs (2008), 251-305). There are 518 protein kinases encoded in the human genome, which were clustered based on their sequence into eight main kinase groups (AGC, CAMK, CK1, CMGC, STE, TK, TKL and Other) in the famous Manning tree (Science (2002), 298, 1912-1934). Each kinase group is further categorized into different kinase families. Since all kinases bind ATP, their binding sites are highly conserved regarding their sequence and structure, making them difficult drug targets when it comes to the development of selective drugs.
Since this protein class is so well-studied, the amount of available data is growing more and more, allowing and requiring infrastructures that organize, analyze, and provide this data to facilitate kinase-centric drug development (Annu. Rep. Med. Chem. (2017), 50, 197-236). One of these rich resources is the KLIFS database, which will be used in this talktorial.
EGFR and Gefitinib¶
The epidermal growth factor receptor (EGFR) is a member of the EGFR (ErbB) family of receptors, which consists of four closely related receptor tyrosine kinases: EGFR (ErbB1), HER2/neu (ErbB2), Her3 (ErbB3) and Her4 (ErbB4). EGFR was the first receptor for which a relationship between mutations, over-expression and cancer has been shown. Interrupting EGFR signaling by blocking the EGFR binding site can help to prevent tumor growth and improve the patient’s health. The first EGFR inhibitor Gefitinib (ligand Expo ID “IRE”) was approved in 2003. (Pharmacol. Res. (2019), 139, 395-411 and Front. Cell Dev. Biol. (2016), 8)
KLIFS database¶
The Kinase-Ligand Interaction Fingerprints and Structures database (KLIFS) is a database that provides information about the protein structure (collected from the PDB) of catalytic kinase domains and the interaction with their ligands.
Role: Kinase-ligand interaction profiles database
Website: http://klifs.net/
API: Yes, REST-based, OpenAPI-enabled. No official client. Uses
bravado.Documentation: http://klifs.net/swagger/
Literature:
Main database/website reference (Nucleic Acids Res. (2020))
Introduction of the KLIFS website & database (Nucleic Acids Res. (2016), 44, 6, D365–D371)
Initial KLIFS dataset, binding mode classification, residue numbering (J. Med. Chem. (2014), 57, 2, 249-277)
Kinase-Ligand Interaction Fingerprints and Structures database (KLIFS), developed at the Division of Medicinal Chemistry - VU University Amsterdam, is a database that revolves around the protein structure of catalytic kinase domains and the way kinase inhibitors can interact with them. Based on the underlying systematic and consistent protocol all (currently human and mouse) kinase structures and the binding mode of kinase ligands can be directly compared to each other. Moreover, because of the classification of an all-encompassing binding site of 85 residues it is possible to compare the interaction patterns of kinase-inhibitors to each other to, for example, identify crucial interactions determining kinase-inhibitor selectivity.
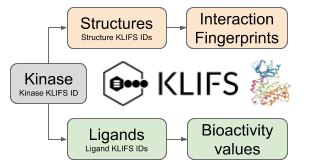
Figure 1: Key kinase resources available on the KLIFS database.
KLIFS OpenAPI¶
The KLIFS database offers standardized URL schemes (REST API) to programmatically access resources that live on the KLIFS server. You could literally paste such an URL into your browser (sending a request) and you would get back a result (receiving a response). For example, the following URL will fetch from KLIFS all ChEMBL bioactivity values associated with the kinase inhibitor Gefitinib (ligand expo ID in the PDB: IRE):
https://klifs.net/api/bioactivity_list_pdb?ligand_PDB=IRE
Now, it would be possible to generate these URL schemes with your own little script or library (client), but it would really be nicer if you would not need to deal with the technical details of how to set up such an URL. Luckily, there is a solution - some websites provide a document that defines the REST API scheme for you (OpenAPI specification, formerly Swagger specification). KLIFS is one of them!
Take a look at how such a document looks like in case of KLIFS (it’s a json file): https://klifs.net/swagger/swagger.json
You can also explore the definitions via an interactive user interface (Swagger UI): http://klifs.net/swagger/
We know now that we can get KLIFS results using URLs and we know thanks to the KLIFS OpenAPI definitions how these URLs need to look like. What we are still missing is a nice Python program (client) that offers a simple Python API to send these requests and receive the responses - under the hood - for us.
Fortunately, libraries like bravado can be used to dynamically generate such a Python client based on OpenAPI definitions. Thus, bravado can be used to generate a KLIFS client based on the KLIFS OpenAPI definitions. This KLIFS client will offer many different methods to interact with the KLIFS webservice (as defined by the OpenAPI definitions). In the Practical section of this talktorial, we will revisit these steps!
opencadd¶
opencadd is a Python library for structural cheminformatics developed by the Volkamer lab. This library is a growing collection of modules that help facilitate and standardize common tasks in structural bioinformatics and cheminformatics, e.g., a module for structural superimposition (opencadd.structure.superposition) or KLIFS queries (opencadd.databases.klifs).
GitHub repository: https://github.com/volkamerlab/opencadd
Documentation: https://opencadd.readthedocs.io
We will use the KLIFS-dedicated module in this talktorial. It offers a standardized API to work with KLIFS data locally (KLIFS download) or remotely (KLIFS OpenAPI). Most query results are returned in the form of standardized pandas DataFrames for quick and easy data manipulation.
Practical¶
[2]:
import logging
logging.getLogger("numexpr").setLevel(logging.ERROR)
from bravado.client import SwaggerClient
from IPython.display import display, Markdown
import pandas as pd
import nglview as nv
from rdkit import Chem
from rdkit.Chem.Draw import IPythonConsole, MolsToGridImage
import opencadd
# Show up to 50 columns for DataFrames in this talktorial
pd.set_option("display.max_columns", 50)
First, we define our kinase and ligand of interest (EGFR and Gefitinib). Next, we explore their KLIFS data using first the KLIFS OpenAPI and then using opencadd’s KLIFS module.
Define kinase and ligand of interest: EGFR and Gefitinib¶
[3]:
species = "Human"
kinase_group = "TK"
kinase_family = "EGFR"
kinase_name = "EGFR"
ligand_expo_id = "IRE"
Generate a KLIFS Python client¶
First, we generate a KLIFS Python client using the provided KLIFS OpenAPI (Swagger) definitions.
[4]:
KLIFS_API_DEFINITIONS = "https://klifs.net/swagger/swagger.json"
KLIFS_CLIENT = SwaggerClient.from_url(KLIFS_API_DEFINITIONS, config={"validate_responses": False})
As with any Python library, you can access the KLIFS client’s entry points when hitting the Tab key after
KLIFS_CLIENT.
Available entry points are:
KLIFS_CLIENT.Information: Access to kinases (name, group, family, …).KLIFS_CLIENT.Interactions: Access to kinase-ligand interaction fingerprints.KLIFS_CLIENT.Ligands: Access to kinase-bound ligands (name, SMILES, …) and their measured bioactivity against kinases (ChEMBL data).KLIFS_CLIENT.Structures: Access to kinase structures with and without ligands (including their KLIFS-processed PDB data).
Explore the KLIFS OpenAPI¶
Let’s send a few requests to KLIFS to explore available data for our kinase and ligand of interest.
1. Kinase groups¶
Let’s check out the available kinase groups.
[5]:
kinase_groups = KLIFS_CLIENT.Information.get_kinase_groups().response().result
# Note: IPython's display function can be used to render output as Markdown
display(Markdown("All kinase groups are returned as a list of strings:"))
print(kinase_groups)
# NBVAL_CHECK_OUTPUT
All kinase groups are returned as a list of strings:
['AGC', 'CAMK', 'CK1', 'CMGC', 'Other', 'STE', 'TK', 'TKL']
2. Kinase families¶
Now let’s look at all available kinase families for the kinase group of interest (kinase_group).
[6]:
# Note: All code lines are supposed to be not longer than 99 characters
# Thus, sometimes brackets are used so that code lines can be split to multiple lines
# (you could also write this in one line without the brackets!)
kinase_families = (
KLIFS_CLIENT.Information.get_kinase_families(kinase_group=kinase_group).response().result
)
display(Markdown(f"Kinase families in {kinase_group} are returned as a list of strings:"))
print(kinase_families)
Kinase families in TK are returned as a list of strings:
['ALK', 'Abl', 'Ack', 'Alk', 'Axl', 'CCK4', 'Csk', 'DDR', 'EGFR', 'Eph', 'FAK', 'FGFR', 'Fer', 'InsR', 'JakA', 'JakB', 'Lmr', 'Met', 'Musk', 'PDGFR', 'Ret', 'Ror', 'Ryk', 'Sev', 'Src', 'Syk', 'TK-Unique', 'Tec', 'Tie', 'Trk', 'VEGFR']
3. Kinases¶
The following kinases belong to the kinase family of interest (kinase_family):
[7]:
kinases = (
KLIFS_CLIENT.Information.get_kinase_names(kinase_family=kinase_family, species=species)
.response()
.result
)
display(
Markdown(
f"Kinases in the {species.lower()} family {kinase_family} as a list of objects "
f"that contain kinase-specific information:"
)
)
kinases
Kinases in the human family EGFR as a list of objects that contain kinase-specific information:
[7]:
[IDlist(full_name='epidermal growth factor receptor', kinase_ID=406, name='EGFR', species='Human'),
IDlist(full_name='erb-b2 receptor tyrosine kinase 2', kinase_ID=407, name='ERBB2', species='Human'),
IDlist(full_name='erb-b2 receptor tyrosine kinase 3', kinase_ID=408, name='ERBB3', species='Human'),
IDlist(full_name='erb-b2 receptor tyrosine kinase 4', kinase_ID=409, name='ERBB4', species='Human')]
In this talktorial, we are interested in the kinase defined in kinase_name. Let’s extract the KLIFS ID for this kinase, which we will use to query structures in KLIFS.
[8]:
# Use iterator to get first element of the list
kinase_klifs_id = next(kinase.kinase_ID for kinase in kinases if kinase.name == kinase_name)
display(Markdown(f"Kinase KLIFS ID for {kinase_name}: {kinase_klifs_id}"))
# NBVAL_CHECK_OUTPUT
Kinase KLIFS ID for EGFR: 406
4. Structures¶
We get all available structures in KLIFS for this kinase and show details for an exemplary ligand-bound structure.
[9]:
structures = (
KLIFS_CLIENT.Structures.get_structures_list(kinase_ID=[kinase_klifs_id]).response().result
)
display(
Markdown(
f"Number of structures for the kinase {kinase_name}: {len(structures)}\n\n"
f"Example structure, i.e. the first structure in the result list that contains a ligand:"
)
)
# If structures do not contain ligands, the ligand field is set to 0
structure = next(structure for structure in structures if structure.ligand != 0)
structure
Number of structures for the kinase EGFR: 565
Example structure, i.e. the first structure in the result list that contains a ligand:
[9]:
structureDetails(DFG='in', Grich_angle=48.1597, Grich_distance=14.8202, Grich_rotation=43.046, aC_helix='out', allosteric_ligand=0, alt='A', back=True, bp_III=False, bp_II_A_in=True, bp_II_B=False, bp_II_B_in=False, bp_II_in=True, bp_II_out=False, bp_IV=False, bp_I_A=True, bp_I_B=True, bp_V=False, chain='A', fp_I=False, fp_II=False, front=True, gate=True, kinase='EGFR', kinase_ID=406, ligand='W19', missing_atoms=0, missing_residues=0, pdb='3w33', pocket='KVLGSGAFGTVYKVAIKELEILDEAYVMASVDPHVCRLLGIQLITQLMPFGCLLDYVREYLEDRRLVHRDLAARNVLVITDFGLA', quality_score=8.0, resolution=1.7, rmsd1=0.814, rmsd2=2.153, species='Human', structure_ID=782)
As you can see for the example structure, KLIFS provides the following information:
the kinase that this structure represents and its pocket sequence
the bound ligand including details on the subpockets that the ligand occupies
the structure quality and conformation
Based on an initial analysis of over 1200 kinase-ligand crystal structures, the KLIFS authors defined a pocket that comprises 85 residues and covers interactions seen in the kinase-ligand structures. For our example, the pocket sequence is the following:
[10]:
display(Markdown(f"Pocket sequence:\n\n{structure.pocket}\n\n({len(structure.pocket)} residues)"))
Pocket sequence:
KVLGSGAFGTVYKVAIKELEILDEAYVMASVDPHVCRLLGIQLITQLMPFGCLLDYVREYLEDRRLVHRDLAARNVLVITDFGLA
(85 residues)
5. Interaction fingerprints¶
Next, we get the interaction fingerprint (IFP) for the example kinase-ligand complex structure. The KLIFS IFP checks for each of the 85 residues and 7 interaction types, if an interaction between that residue and the ligand is available (1) or not (0).
[11]:
KLIFS_CLIENT.Interactions.get_interactions_get_types().response().result
[11]:
[{'position': '1', 'name': 'Apolar contact'},
{'position': '2', 'name': 'Aromatic face-to-face'},
{'position': '3', 'name': 'Aromatic edge-to-face'},
{'position': '4', 'name': 'Hydrogen bond donor (protein)'},
{'position': '5', 'name': 'Hydrogen bond acceptor (protein)'},
{'position': '6', 'name': 'Protein cation - ligand anion'},
{'position': '7', 'name': 'Protein anion - ligand cation'}]
This results in an IFP with \(85 \cdot 7 = 595\) bits. Let’s look at the IFP for our example structure.
[12]:
structure_klifs_id = structure.structure_ID
interaction_fingerprints = (
KLIFS_CLIENT.Interactions.get_interactions_get_IFP(structure_ID=[structure_klifs_id])
.response()
.result
)
interaction_fingerprints
[12]:
[{'structure_ID': 782,
'IFP': '0000000000000010000001000000000000000000000000000000000000000000000000100000000000000000000000000010000001000000100000000000000000000000000000000000000000000000000000000000000000000000000001000000000000000000000000000000000000000000000000000000010000001000000100000000000000000000000000000000001000000100000010000001000000100000010011000000000000000010000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000100000000000000000000100000010000001010000000000010000000000000'}]
Further below in this talktorial, we will show a quick example of how you could use this IFP information.
6. Structure coordinates¶
We explored so far a lot the structure properties. Let’s get the actual structural data for this first structure in the form of a string that contains the atomic coordinates for the kinase-ligand complex.
[13]:
complex_coordinates = (
KLIFS_CLIENT.Structures.get_structure_get_pdb_complex(structure_ID=structures[0].structure_ID)
.response()
.result
)
# Show some example rows
complex_coordinates.split("\n")[100:110]
[13]:
['ATOM 67 N GLU A 709 18.017 26.733 45.799 1.00 0.00 N ',
'ATOM 68 CA GLU A 709 17.685 25.527 46.562 1.00 0.00 C ',
'ATOM 69 C GLU A 709 17.372 25.814 48.036 1.00 0.00 C ',
'ATOM 70 O GLU A 709 16.634 25.071 48.672 1.00 0.00 O ',
'ATOM 71 CB GLU A 709 18.816 24.503 46.474 1.00 0.00 C ',
'ATOM 72 CG GLU A 709 18.851 23.724 45.177 1.00 0.00 C ',
'ATOM 73 CD GLU A 709 19.877 22.613 45.199 1.00 0.00 C ',
'ATOM 74 OE1 GLU A 709 19.470 21.427 45.212 1.00 0.00 O ',
'ATOM 75 OE2 GLU A 709 21.088 22.925 45.212 1.00 0.00 O1-',
'ATOM 76 N THR A 710 17.928 26.893 48.572 1.00 0.00 N ']
7. Ligands¶
Last but not least, let’s have a quick look at another important resource in KLIFS. For each ligand, it is possible to extract all bioactivities measured against different kinases. The bioactivity data comes from ChEMBL, a manually curated compound database (check out Talktorial T001 for more information about ChEMBL).
[14]:
bioactivities = (
KLIFS_CLIENT.Ligands.get_bioactivity_list_pdb(ligand_PDB=structure.ligand).response().result
)
display(
Markdown(
f"Number of ChEMBL bioactivity values available in KLIFS for example ligand {structure.ligand}: {len(bioactivities)}\n\n"
f"Example bioactivity:"
)
)
bioactivities[0]
Number of ChEMBL bioactivity values available in KLIFS for example ligand W19: 10
Example bioactivity:
[14]:
{'pref_name': 'Dual specificity mitogen-activated protein kinase kinase 5',
'accession': 'Q13163',
'organism': 'Homo sapiens',
'standard_type': 'IC50',
'standard_relation': '=',
'standard_value': '870.0',
'standard_units': 'nM',
'pchembl_value': '6.06'}
Case study: EGFR (using opencadd)¶
After introducing the KLIFS OpenAPI above, we use now the opencadd.databases.klifs module, which is a wrapper for this KLIFS client. As mentioned in the Theory section of this talktorial, this module returns all responses as pandas DataFrames for easy and quick manipulation.
[15]:
from opencadd.databases.klifs import setup_remote
session = setup_remote()
1. Get all structures for EGFR¶
[16]:
structures = session.structures.by_kinase_klifs_id(kinase_klifs_id)
display(Markdown(f"Number of structures for the kinase {kinase_name}: {len(structures)}"))
Number of structures for the kinase EGFR: 565
Get IFPs for the queried EGFR structures. Note that only ligand-bound structures can have an IFP.
[17]:
structure_klifs_ids = structures["structure.klifs_id"].to_list()
interaction_fingerprints = session.interactions.by_structure_klifs_id(structure_klifs_ids)
display(
Markdown(
f"Number of IFPs for {kinase_name}: {len(interaction_fingerprints)}\n\n"
f"Show example IFPs:"
)
)
interaction_fingerprints.head()
Number of IFPs for EGFR: 508
Show example IFPs:
[17]:
| structure.klifs_id | interaction.fingerprint | |
|---|---|---|
| 0 | 775 | 0000000000000010000000000000000000000000000000... |
| 1 | 777 | 0000000000000010000001000000000000000000000000... |
| 2 | 778 | 0000000000000010000000000000000000000000000000... |
| 3 | 779 | 0000000000000010000001000000000000000000000000... |
| 4 | 781 | 0000000000000010000001000000000000000000000000... |
2. Average interaction fingerprint¶
Let’s aggregate the information about interaction types per residue positions from all ligand-bound structures that are available for our kinase of interest.
[18]:
def average_n_interactions_per_residue(structure_klifs_ids):
"""
Generate residue position x interaction type matrix that contains
the average number of interactions per residue and interaction type.
Parameters
----------
structure_klifs_ids : list of int
Structure KLIFS IDs.
Returns
-------
pandas.DataFrame
Average number of interactions per residue (rows) and interaction type (columns).
"""
# Get IFP (is returned from KLIFS as string)
ifps = session.interactions.by_structure_klifs_id(structure_klifs_ids)
# Split string into list of int (0, 1): structures x IFP bits matrix
ifps = pd.DataFrame(ifps["interaction.fingerprint"].apply(lambda x: list(x)).to_list())
ifps = ifps.astype("int32")
# Sum up all interaction per bit position and normalize by number of IFPs
ifp_relative = (ifps.sum() / len(ifps)).to_list()
# Transform aggregated IFP into residue position x interaction type matrix
residue_feature_matrix = pd.DataFrame(
[ifp_relative[i : i + 7] for i in range(0, len(ifp_relative), 7)], index=range(1, 86)
)
# Add interaction type names
columns = session.interactions.interaction_types["interaction.name"].to_list()
residue_feature_matrix.columns = columns
return residue_feature_matrix
[19]:
residue_feature_matrix = average_n_interactions_per_residue(structure_klifs_ids)
display(
Markdown(
f"Calculated average interactions per interaction type (feature) and residue position:\n\n"
f"**Interaction types** (columns): {', '.join(residue_feature_matrix.columns)}\n\n"
f"**Residue positions** (rows): {residue_feature_matrix.index}"
)
)
# NBVAL_CHECK_OUTPUT
Calculated average interactions per interaction type (feature) and residue position:
Interaction types (columns): Apolar contact, Aromatic face-to-face, Aromatic edge-to-face, Hydrogen bond donor (protein), Hydrogen bond acceptor (protein), Protein cation - ligand anion, Protein anion - ligand cation
Residue positions (rows): RangeIndex(start=1, stop=86, step=1)
[20]:
residue_feature_matrix.head()
[20]:
| Apolar contact | Aromatic face-to-face | Aromatic edge-to-face | Hydrogen bond donor (protein) | Hydrogen bond acceptor (protein) | Protein cation - ligand anion | Protein anion - ligand cation | |
|---|---|---|---|---|---|---|---|
| 1 | 0.001969 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 |
| 2 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 |
| 3 | 0.976378 | 0.0 | 0.0 | 0.0 | 0.003937 | 0.0 | 0.0 |
| 4 | 0.679134 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 |
| 5 | 0.322835 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 |
[21]:
residue_feature_matrix.plot.bar(
figsize=(15, 5),
stacked=True,
xlabel="KLIFS pocket residue position",
ylabel="Average number of interactions (per interaction type)",
color=["grey", "green", "limegreen", "red", "blue", "orange", "cyan"],
);
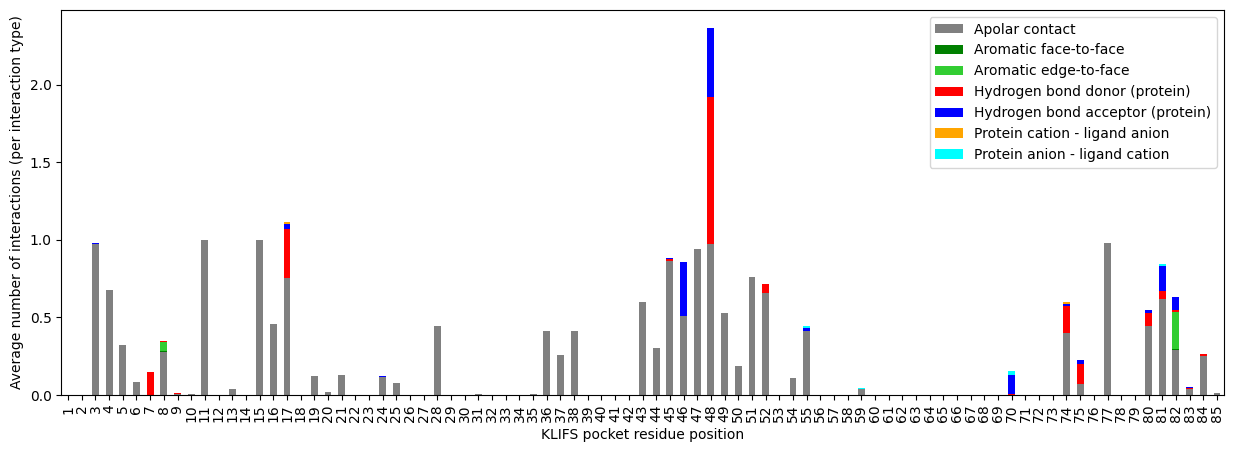
The apolar contacts nicely show which residues are in close contact with the hydrophobic parts of the ligands. Most ligands show hydrogen bonding at position 48, which is part of the binding site’s hinge region, a hydrophilic anchor position for kinase-ligand binding.
3. Select an example EGFR-Gefitinib structure¶
Keep only structures bound to a ligand of interest (ligand_expo_id), sort them by highest resolution and show the top 10 structures.
[22]:
subset = structures[(structures["ligand.expo_id"] == ligand_expo_id)]
subset.sort_values(by="structure.resolution").head(10)
[22]:
| structure.klifs_id | structure.pdb_id | structure.alternate_model | structure.chain | species.klifs | kinase.klifs_id | kinase.klifs_name | kinase.names | kinase.family | kinase.group | structure.pocket | ligand.expo_id | ligand_allosteric.expo_id | ligand.klifs_id | ligand_allosteric.klifs_id | ligand.name | ligand_allosteric.name | structure.dfg | structure.ac_helix | structure.resolution | structure.qualityscore | structure.missing_residues | structure.missing_atoms | structure.rmsd1 | structure.rmsd2 | interaction.fingerprint | structure.front | structure.gate | structure.back | structure.fp_i | structure.fp_ii | structure.bp_i_a | structure.bp_i_b | structure.bp_ii_in | structure.bp_ii_a_in | structure.bp_ii_b_in | structure.bp_ii_out | structure.bp_ii_b | structure.bp_iii | structure.bp_iv | structure.bp_v | structure.grich_distance | structure.grich_angle | structure.grich_rotation | structure.filepath | structure.curation_flag | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 42 | 823 | 4i22 | - | A | Human | 406 | EGFR | <NA> | <NA> | <NA> | KVLGSGAFGTVYKVAIKELEILDEAYVMASVDPHVCRLLGIQLIMQ... | IRE | - | 222 | 0 | <NA> | <NA> | in | out | 1.71 | 8.0 | 0 | 0 | 0.810 | 2.159 | <NA> | True | True | False | False | False | True | True | False | False | False | False | False | False | False | False | 14.6390 | 47.539398 | 47.973499 | <NA> | False |
| 47 | 837 | 4wkq | B | A | Human | 406 | EGFR | <NA> | <NA> | <NA> | KVLGSG___TVYKVAIKE_EILDEAYVMASVDPHVCRLLGIQLITQ... | IRE | - | 222 | 0 | <NA> | <NA> | in | in | 1.85 | 6.7 | 5 | 13 | 0.776 | 1.995 | <NA> | True | True | False | False | False | True | True | False | False | False | False | False | False | False | False | 0.0000 | 0.000000 | 0.000000 | <NA> | False |
| 93 | 889 | 4wkq | A | A | Human | 406 | EGFR | <NA> | <NA> | <NA> | KVLGSG___TVYKVAIKE_EILDEAYVMASVDPHVCRLLGIQLITQ... | IRE | - | 222 | 0 | <NA> | <NA> | in | in | 1.85 | 6.7 | 5 | 13 | 0.776 | 1.995 | <NA> | True | True | False | False | False | True | True | False | False | False | False | False | False | False | False | 0.0000 | 0.000000 | 0.000000 | <NA> | False |
| 23 | 819 | 3ug2 | B | A | Human | 406 | EGFR | <NA> | <NA> | <NA> | KVLSSG___TVYKVAIKE_EILDEAYVMASVDPHVCRLLGIQLIMQ... | IRE | - | 222 | 0 | <NA> | <NA> | in | in | 2.50 | 7.6 | 4 | 8 | 0.778 | 2.014 | <NA> | True | True | True | False | False | True | True | False | False | False | False | False | False | False | False | 0.0000 | 0.000000 | 0.000000 | <NA> | False |
| 98 | 898 | 3ug2 | A | A | Human | 406 | EGFR | <NA> | <NA> | <NA> | KVLSSG___TVYKVAIKE_EILDEAYVMASVDPHVCRLLGIQLIMQ... | IRE | - | 222 | 0 | <NA> | <NA> | in | in | 2.50 | 7.6 | 4 | 8 | 0.778 | 2.014 | <NA> | True | True | True | False | False | True | True | False | False | False | False | False | False | False | False | 0.0000 | 0.000000 | 0.000000 | <NA> | False |
| 18 | 789 | 2itz | - | A | Human | 406 | EGFR | <NA> | <NA> | <NA> | KVLGSGAFGTVYKVAIKELEILDEAYVMASVDPHVCRLLGIQLITQ... | IRE | - | 222 | 0 | <NA> | <NA> | in | in | 2.72 | 9.4 | 0 | 6 | 0.776 | 2.097 | <NA> | True | True | False | False | False | True | True | False | False | False | False | False | False | False | False | 18.0567 | 58.131699 | 30.073000 | <NA> | False |
| 2 | 783 | 2ito | - | A | Human | 406 | EGFR | <NA> | <NA> | <NA> | KVLSSGAFGTVYKVAIKELEILDEAYVMASVDPHVCRLLGIQLITQ... | IRE | - | 222 | 0 | <NA> | <NA> | in | in | 3.25 | 8.0 | 0 | 0 | 0.778 | 2.096 | <NA> | True | True | False | False | False | True | True | False | False | False | False | False | False | False | False | 19.7766 | 63.316700 | 23.749100 | <NA> | False |
| 20 | 790 | 2ity | - | A | Human | 406 | EGFR | <NA> | <NA> | <NA> | KVLGSGAFGTVYKVAIKELEILDEAYVMASVDPHVCRLLGIQLITQ... | IRE | - | 222 | 0 | <NA> | <NA> | in | in | 3.42 | 8.0 | 0 | 0 | 0.778 | 2.103 | <NA> | True | True | False | False | False | True | True | False | False | False | False | False | False | False | False | 19.5474 | 62.976700 | 21.934299 | <NA> | False |
[23]:
structure_klifs_id = (
structures[(structures["ligand.expo_id"] == ligand_expo_id)]
.sort_values(by="structure.resolution")
.iloc[0]["structure.klifs_id"]
)
message = f"Structure KLIFS ID for best resolved {ligand_expo_id}-bound {kinase_name}: {structure_klifs_id}"
display(Markdown(message))
Structure KLIFS ID for best resolved IRE-bound EGFR: 823
4. Show the structure with nglview¶
Let’s preview the protein-ligand complex with nglview.
[24]:
def show_molecule(structure_text, extension="mol2"):
"""
Show molecule with nglview.
Parameters
----------
structure_text : str
Structural data in the form of a string.
extension : str
Format of the structural data (default: mol2).
Returns
-------
nglview.widget.NGLWidget
Molecular viewer.
"""
v = nv.NGLWidget()
v.add_component(structure_text, ext=extension)
return v
[25]:
structure_text = session.coordinates.to_text(structure_klifs_id, "complex", "pdb")
v = show_molecule(structure_text, "pdb")
v
[26]:
v.render_image();
[27]:
v._display_image()
[27]:
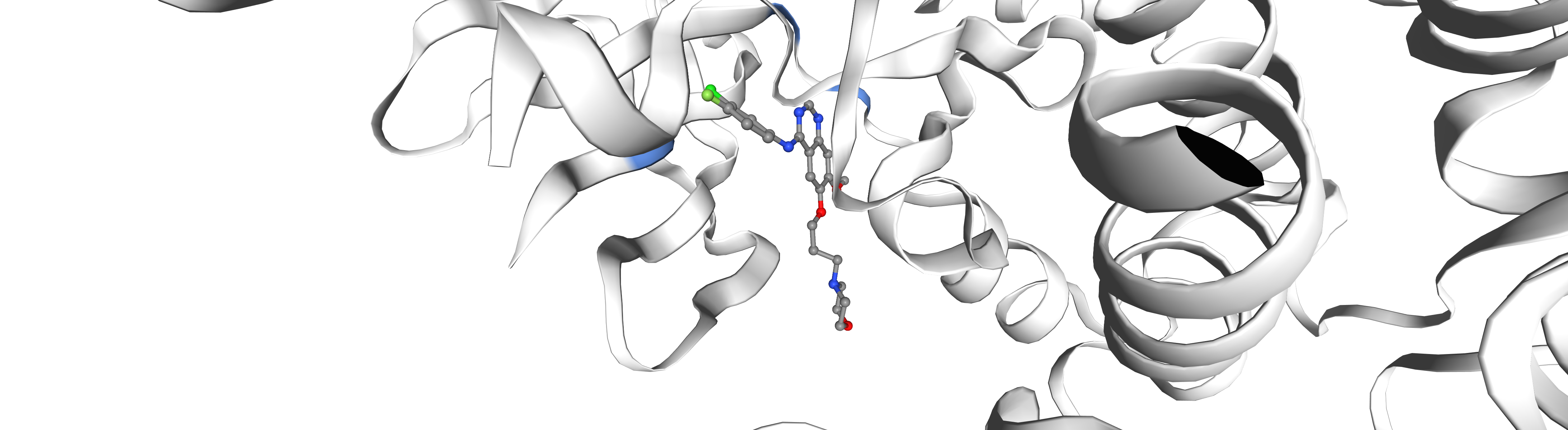
Let’s highlight residues with frequent interactions (across all IFPs for a kinase with kinase_klifs_id, in our case kinase EGFR) in an example structure with structure_klifs_id. Define with features, which feature types you would like to look at, and with feature_cutoff, which cutoff to use for highlighting (None means you will see any residue that has shown at least one interaction across all structures).
[28]:
def show_kinase_feature_positions(
kinase_klifs_id, structure_klifs_id, features=["HBD"], feature_cutoff=None
):
"""
Show an example kinase structure and color residues showing interactions of defined type(s)
across interaction fingerprints for this kinase.
Parameters
----------
kinase_klifs_id : int
Kinase KLIFS ID used to fetch all associated IFPs.
structure_klifs_id : int
Structure KLIFS ID used to map average feature coverage per residue.
features : list of str
One or more feature types (H, AR.f2f, AR.e2f, HBD, HBA, NI, PI).
feature_cutoff : int or float
Show only residues with a higher average feature coverage than this cutoff.
Returns
-------
nglview.widget.NGLWidget
Molecular viewer.
"""
# Get all structures KLIFS ID for a kinase
structures = session.structures.by_kinase_klifs_id(kinase_klifs_id)
structure_klifs_ids = structures["structure.klifs_id"].to_list()
# Get aggregated residue-feature matrix from IFPs
residue_feature_matrix = average_n_interactions_per_residue(structure_klifs_ids)
residue_feature_matrix.columns = "H AR.f2f AR.e2f HBD HBA NI PI".split()
# Aggregate feature values
residues_features = residue_feature_matrix[features].sum(axis=1)
# Fetch the PDB residue numbers for the 85 pocket residues
index = session.pockets.by_structure_klifs_id(structure_klifs_id)["residue.id"]
residues_features.index = index
# Keep only residues with feature occurrence above a cutoff occurrence
if feature_cutoff:
residues_features = residues_features[residues_features > feature_cutoff]
# Prepare residue-color mapping in a nglview-required format
color_scheme_list = [
["cornflowerblue", residue_id] for residue_id, _ in residues_features.items()
]
color_scheme = nv.color._ColorScheme(color_scheme_list, label="interactions")
structure_text = session.coordinates.to_text(structure_klifs_id, "complex", "pdb")
ligand_expo_id = session.structures.by_structure_klifs_id(structure_klifs_id).iloc[0][
"ligand.expo_id"
]
v = show_molecule(structure_text, "pdb")
v.clear()
v.add_representation("cartoon", selection="protein", color=color_scheme)
v.add_representation("ball+stick", selection=ligand_expo_id)
return v
[29]:
selected_features = ["HBD", "HBA"]
feature_cutoff = 0.25
[30]:
display(
Markdown(
f"Show residues with interaction(s) {selected_features} for kinase #{kinase_klifs_id} "
f"on structure #{structure_klifs_id} that have been seen in at least {feature_cutoff} of all structures."
)
)
v = show_kinase_feature_positions(
kinase_klifs_id, structure_klifs_id, features=selected_features, feature_cutoff=0.25
)
v.center(selection=ligand_expo_id)
v
Show residues with interaction(s) [‘HBD’, ‘HBA’] for kinase #406 on structure #823 that have been seen in at least 0.25 of all structures.
[31]:
v.render_image();
[32]:
v._display_image()
[32]:
5. Show all kinase-bound ligands with rdkit¶
Let’s take a look at all EGFR-bound ligands using rdkit, a popular library for cheminformatics.
[33]:
def show_ligands(smiles):
return Chem.Draw.MolsToGridImage(
[Chem.MolFromSmiles(s) for s in smiles], maxMols=10, molsPerRow=5
)
[34]:
ligands = session.ligands.by_kinase_klifs_id(kinase_klifs_id)
display(Markdown(f"Number of ligands bound to kinase #{kinase_klifs_id}: {len(ligands)}"))
# Show only the first 10 ligands
show_ligands(ligands["ligand.smiles"].to_list()[:10])
Number of ligands bound to kinase #406: 153
[34]:
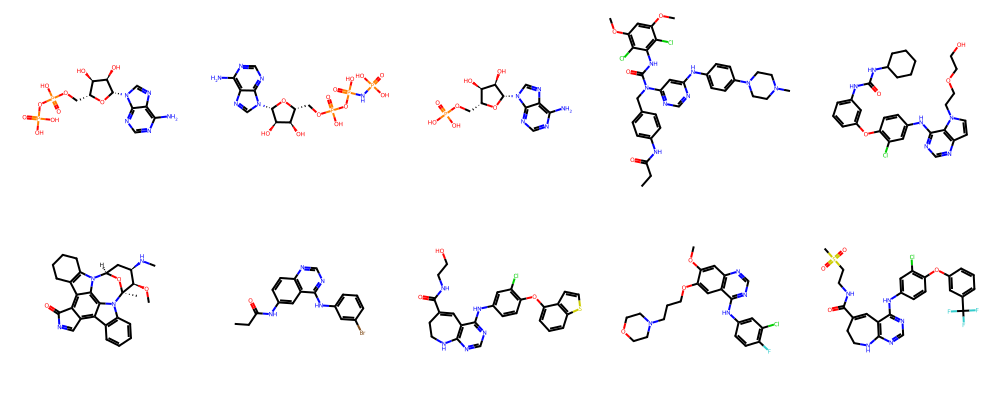
6. Explore profiling data for Gefitinib¶
Get profiling data (bioactivities) for Gefitinib.
[35]:
bioactivities = session.bioactivities.by_ligand_expo_id(ligand_expo_id)
display(
Markdown(
f"Number of bioactivity values for {ligand_expo_id}: {len(bioactivities)}\n\n"
f"Show example bioactivities:\n\n"
)
)
bioactivities.sort_values("ligand.bioactivity_standard_value").head()
Number of bioactivity values for IRE: 338
Show example bioactivities:
[35]:
| kinase.pref_name | kinase.uniprot | kinase.chembl_id | ligand.chembl_id | ligand.bioactivity_standard_type | ligand.bioactivity_standard_relation | ligand.bioactivity_standard_value | ligand.bioactivity_standard_units | ligand.bioactivity_pchembl_value | species.chembl | ligand.expo_id (query) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 60 | Epidermal growth factor receptor erbB1 | P00533 | CHEMBL203 | CHEMBL939 | IC50 | = | 0.10 | nM | 10.00 | Homo sapiens | IRE |
| 106 | Epidermal growth factor receptor erbB1 | P00533 | CHEMBL203 | CHEMBL939 | IC50 | = | 0.15 | nM | 9.82 | Homo sapiens | IRE |
| 107 | Epidermal growth factor receptor erbB1 | P00533 | CHEMBL203 | CHEMBL939 | IC50 | = | 0.17 | nM | 9.77 | Homo sapiens | IRE |
| 62 | Epidermal growth factor receptor erbB1 | P00533 | CHEMBL203 | CHEMBL939 | IC50 | = | 0.20 | nM | 9.70 | Homo sapiens | IRE |
| 154 | Epidermal growth factor receptor erbB1 | P00533 | CHEMBL203 | CHEMBL939 | IC50 | = | 0.32 | nM | 9.49 | Homo sapiens | IRE |
Show the bioactivity distribution!
[36]:
bioactivities["ligand.bioactivity_standard_value"].plot(
kind="hist", title=f"ChEMBL bioactivity standard values (nM) for {ligand_expo_id}"
);
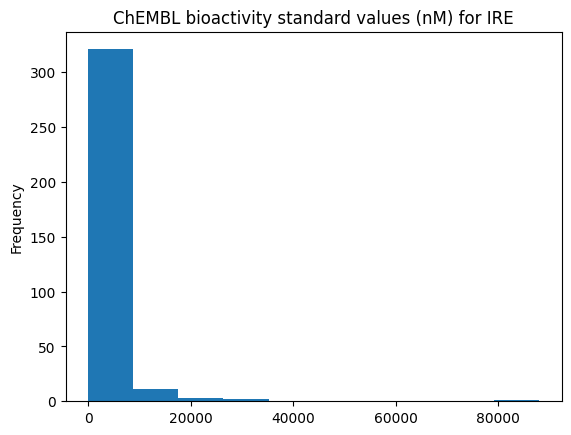
We can see that there are many measurements with high bioactivity (low values) and some with no bioactivity (high values). We know that the intended target (on-target) of Gefitinib is EGFR (also called epidermal growth factor receptor erbB1). It would be interesting to see if there are other kinases that have been shown to bind Gefitinib strongly (off-targets). Such off-targets are often the reason for side effects of drugs.
In order to find off-targets based on the ChEMBL profiling data at hand, we filter our bioactivity dataset only for measurements showing high activity using the cutoff activity_cutoff and we will print out all measurements for targets that are not EGFR, i.e. potential off-targets.
[37]:
ACTIVITY_CUTOFF = 100
bioactivities_active = bioactivities[
bioactivities["ligand.bioactivity_standard_value"] < ACTIVITY_CUTOFF
]
display(Markdown("Number of measurements with high activity per kinase:"))
n_bioactivities_per_target = (
bioactivities_active.groupby("kinase.pref_name").size().sort_values(ascending=True)
)
n_bioactivities_per_target
Number of measurements with high activity per kinase:
[37]:
kinase.pref_name
Interleukin-1 receptor-associated kinase 1 1
Nuclear factor NF-kappa-B p65 subunit 1
Vascular endothelial growth factor receptor 2 1
Serine/threonine-protein kinase RIPK2 1
Serine/threonine-protein kinase GAK 2
Receptor protein-tyrosine kinase erbB-2 3
Epidermal growth factor receptor erbB1 99
dtype: int64
[38]:
display(Markdown(f"Off-targets of {ligand_expo_id} based on profiling data:"))
bioactivities_active[
bioactivities_active["kinase.pref_name"] != "Epidermal growth factor receptor erbB1"
].sort_values(["ligand.bioactivity_standard_value"])
Off-targets of IRE based on profiling data:
[38]:
| kinase.pref_name | kinase.uniprot | kinase.chembl_id | ligand.chembl_id | ligand.bioactivity_standard_type | ligand.bioactivity_standard_relation | ligand.bioactivity_standard_value | ligand.bioactivity_standard_units | ligand.bioactivity_pchembl_value | species.chembl | ligand.expo_id (query) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 277 | Serine/threonine-protein kinase RIPK2 | O43353 | CHEMBL5014 | CHEMBL939 | IC50 | = | 3.800000 | nM | 8.42 | Homo sapiens | IRE |
| 272 | Serine/threonine-protein kinase GAK | O14976 | CHEMBL4355 | CHEMBL939 | Kd | = | 7.000000 | nM | 8.15 | Homo sapiens | IRE |
| 270 | Serine/threonine-protein kinase GAK | O14976 | CHEMBL4355 | CHEMBL939 | Kd | = | 13.000000 | nM | 7.89 | Homo sapiens | IRE |
| 241 | Receptor protein-tyrosine kinase erbB-2 | P04626 | CHEMBL1824 | CHEMBL939 | IC50 | = | 24.000000 | nM | 7.62 | Homo sapiens | IRE |
| 246 | Receptor protein-tyrosine kinase erbB-2 | P04626 | CHEMBL1824 | CHEMBL939 | Ki | = | 25.120001 | nM | 7.60 | Homo sapiens | IRE |
| 335 | Vascular endothelial growth factor receptor 2 | P35968 | CHEMBL279 | CHEMBL939 | IC50 | = | 48.500000 | nM | 7.31 | Homo sapiens | IRE |
| 219 | Nuclear factor NF-kappa-B p65 subunit | Q04206 | CHEMBL5533 | CHEMBL939 | IC50 | = | 55.000000 | nM | 7.26 | Homo sapiens | IRE |
| 194 | Interleukin-1 receptor-associated kinase 1 | P51617 | CHEMBL3357 | CHEMBL939 | Kd | = | 69.000000 | nM | 7.16 | Homo sapiens | IRE |
| 238 | Receptor protein-tyrosine kinase erbB-2 | P04626 | CHEMBL1824 | CHEMBL939 | IC50 | = | 97.000000 | nM | 7.01 | Homo sapiens | IRE |
Let’s take a look at the off-targets and see if we can rationalize our results from a kinase similarity point of view (similar kinases are likely to bind the same ligand) - we will focus on sequence similarity (aka kinase groups and families) for now.
The off-target list contains closely related kinases:
from the tyrosine kinase (TK) group, to which EGFR itself belongs: e.g. ErbB2 (another member of the EGFR family) and VEGFR/KDR
from the related tyrosine-like kinase (TKL) group: e.g. RIPK2 and IRAK1
We also find the GAK kinase, which belongs to the atypical kinases and might not be an obvious off-target when only looking at sequence similarity, but it is another well-known and reported off-target. This is a nice example of how sequence is not the only indicator of protein similarity but that structural similarity plays a very important role as well.
Check out Talktorial T010 for more details on binding site similarity and off-target prediction.
Explore a random kinase in KLIFS¶
This section is intended to quickly explore the resources on different kinases in KLIFS.
First, we define a function that allows us to return (a) all structure-bound ligands in KLIFS plus (b) one random structure for a random kinase or given kinase(s).
[39]:
def get_random_kinase_structure_with_ligand_information(*kinase_names, seed=None):
"""
Get one example structure (complex and protein structural data) for one or more kinases
plus the SMILES of all bound ligands.
Parameters
----------
*kinase_names : str
Kinase names.
seed : int, array-like, BitGenerator, np.random.RandomState
Seed for random number generator. Default is None.
Returns
-------
molcomplex : str
Complex structural data.
protein : str
Protein structural data.
ligands : list of str
List of ligand SMILES.
"""
# Get all structures for a given kinase
if kinase_names:
kinase_names = list(kinase_names)
try:
structures = session.structures.by_kinase_name(list(kinase_names))
except ValueError:
raise ValueError(f"The input kinase(s) {kinase_names} yielded no results.")
# Or get all available structures
else:
structures = session.structures.all_structures()
kinase_klifs_ids = [int(i) for i in structures["kinase.klifs_id"].unique()]
# Choose random structure and corresponding kinase
structure = structures.sample(random_state=seed).squeeze()
structure_klifs_id = structure["structure.klifs_id"]
kinase_klifs_id = structure["kinase.klifs_id"]
display(Markdown(f"Selected kinases: {structure['kinase.klifs_name']}"))
# Get complex structural data
molcomplex = (
KLIFS_CLIENT.Structures.get_structure_get_pdb_complex(structure_ID=structure_klifs_id)
.response()
.result
)
# Get protein structural data
protein = (
KLIFS_CLIENT.Structures.get_structure_get_protein(structure_ID=structure_klifs_id)
.response()
.result
)
# Get all ligands bound to all structures for input kinases
ligands = KLIFS_CLIENT.Ligands.get_ligands_list(kinase_ID=[kinase_klifs_id]).response().result
display(
Markdown(
f"Chosen KLIFS entry: PDB ID {structure['structure.pdb_id']} "
f"with chain {structure['structure.chain'] if structure['structure.chain'] else '-'} and "
f"alternate model {structure['structure.alternate_model'] if structure['structure.alternate_model'] else '-'} "
f"({structure['species.klifs'].lower()}).\n\n"
f"Number of structure-bound ligands: {len(ligands)}"
)
)
return molcomplex, protein, [ligand.SMILES for ligand in ligands]
Second, we use the function to get data on a random kinase. Note: You can specify one or more kinases of your choice to narrow down the search space if you want, see random_kinase_structure(*kinase_names).
[40]:
# We are setting a seed here for reproducible results
# Remove the seed to explore random kinases
molcomplex, protein, ligands = get_random_kinase_structure_with_ligand_information(seed=1)
Selected kinases: DRAK2
Chosen KLIFS entry: PDB ID 6y6f with chain A and alternate model A (human).
Number of structure-bound ligands: 8
[41]:
show_ligands(ligands[:10])
[41]:
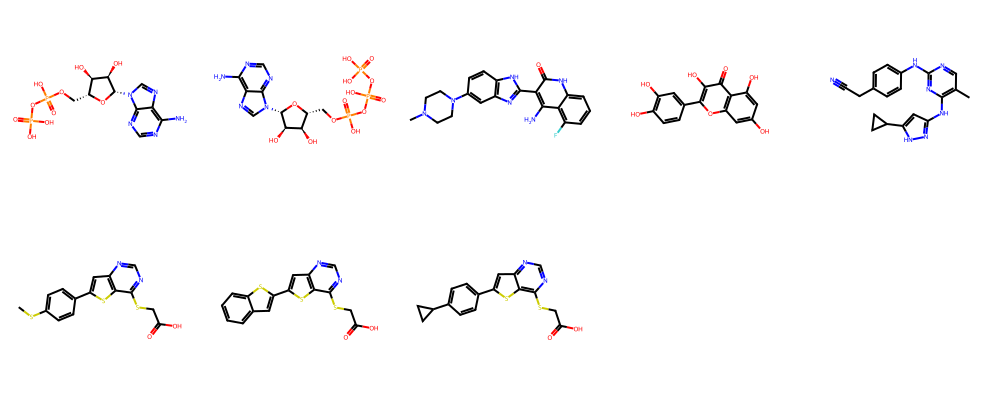
[42]:
v = show_molecule(molcomplex, "pdb")
v
[43]:
v.render_image();
[45]:
v._display_image()
[45]:
Discussion¶
In this talktorial, we have explored how to interact with the rich kinase resource KLIFS using (a) the KLIFS OpenAPI and (b) the KLIFS-dedicated module in opencadd. First, we have used the kinase EGFR to extract all available ligand-bound structures and their KLIFS interaction fingerprints, which were used to extract key hydrogen bonding residues. Second, we fetched profiling data for Gefitinib to investigate EGFR off-targets.
Quiz¶
Explain and set in context the following terms: Server, client, API, request, response, REST and OpenAPI.
Query KLIFS: How many kinases does KLIFS provide for the Abl family? How many structures are available for the kinase CDK2?
Explain how the profiling data of an inhibitor can be used to search for off-targets.
[ ]: